dotan1111/MSA-nuc-10-seq
收藏Hugging Face2023-09-18 更新2024-03-04 收录
下载链接:
https://hf-mirror.com/datasets/dotan1111/MSA-nuc-10-seq
下载链接
链接失效反馈官方服务:
资源简介:
---
tags:
- sequence-to-sequence
- bioinformatics
- biology
---
# Multiple Sequence Alignment as a Sequence-to-Sequence Learning Problem
## Abstract:
The sequence alignment problem is one of the most fundamental problems in bioinformatics and a plethora of methods were devised to tackle it. Here we introduce BetaAlign, a methodology for aligning sequences using an NLP approach. BetaAlign accounts for the possible variability of the evolutionary process among different datasets by using an ensemble of transformers, each trained on millions of samples generated from a different evolutionary model. Our approach leads to alignment accuracy that is similar and often better than commonly used methods, such as MAFFT, DIALIGN, ClustalW, T-Coffee, PRANK, and MUSCLE.
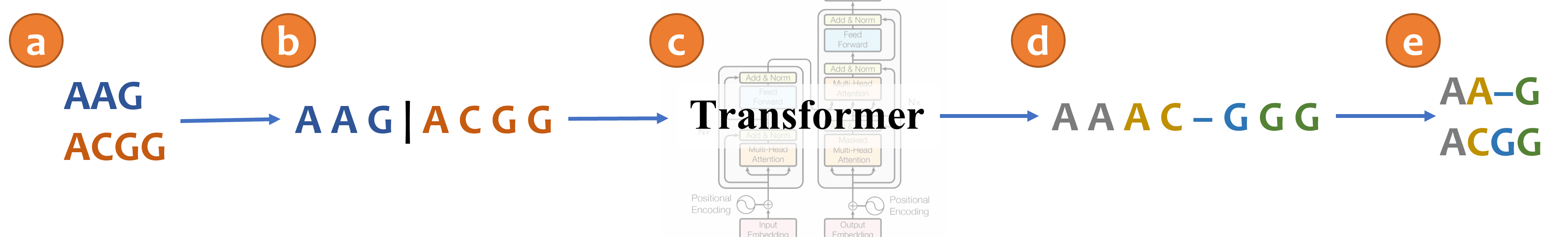
An illustration of aligning sequences with sequence-to-sequence learning. (a) Consider two input sequences "AAG" and "ACGG". (b) The result of encoding the unaligned sequences into the source language (*Concat* representation). (c) The sentence from the source language is translated to the target language via a transformer model. (d) The translated sentence in the target language (*Spaces* representation). (e) The resulting alignment, decoded from the translated sentence, in which "AA-G" is aligned to "ACGG". The transformer architecture illustration is adapted from (Vaswani et al., 2017).
## Data:
We used SpartaABC (Loewenthal et al., 2021) to generate millions of true alignments. SpartaABC requires the following input: (1) a rooted phylogenetic tree, which includes a topology and branch lengths; (2) a substitution model (amino acids or nucleotides); (3) root sequence length; (4) the indel model parameters, which include: insertion rate (*R_I*), deletion rate (*R_D*), a parameter for the insertion Zipfian distribution (*A_I*), and a parameter for the deletion Zipfian distribution (*A_D*). MSAs were simulated along random phylogenetic tree topologies generated using the program ETE version 3.0 (Huerta-Cepas et al., 2016) with default parameters.
We generated 1,495,000, 2,000 and 3,000, protein MSAs with ten sequences that were used as training validation and testing data, respectively. We generated the same number of DNA MSAs. For each random tree, branch lengths were drawn from a uniform distribution in the range *(0.5,1.0)*. Next, the sequences were generated using SpartaABC with the following parameters: *R_I,R_D \in (0.0,0.05)*, *A_I, A_D \in (1.01,2.0)*. The alignment lengths as well as the sequence lengths of the tree leaves vary within and among datasets as they depend on the indel dynamics and the root length. The root length was sampled uniformly in the range *[32,44]*. Unless stated otherwise, all protein datasets were generated with the WAG+G model, and all DNA datasets were generated with the GTR+G model, with the following parameters: (1) frequencies for the different nucleotides *(0.37, 0.166, 0.307, 0.158)*, in the order "T", "C", "A" and "G"; (2) with the substitutions rate *(0.444, 0.0843, 0.116, 0.107, 0.00027)*, in the order "a", "b", "c", "d", and "e" for the substitution matrix.
## Example:
The following example correspond for the illustrated MSA in the figure above:
{"MSA": "AAAC-GGG", "unaligned_seqs": {"seq0": "AAG", "seq1": "ACGG"}}
## APA
```
Dotan, E., Belinkov, Y., Avram, O., Wygoda, E., Ecker, N., Alburquerque, M., Keren, O., Loewenthal, G., & Pupko T. (2023). Multiple sequence alignment as a sequence-to-sequence learning problem. The Eleventh International Conference on Learning Representations (ICLR 2023).
```
## BibTeX
```
@article{Dotan_multiple_2023,
author = {Dotan, Edo and Belinkov, Yonatan and Avram, Oren and Wygoda, Elya and Ecker, Noa and Alburquerque, Michael and Keren, Omri and Loewenthal, Gil and Pupko, Tal},
month = aug,
title = {{Multiple sequence alignment as a sequence-to-sequence learning problem}},
year = {2023}
}
```
The dataset is generated by the BetaAlign method, which addresses the sequence alignment problem in bioinformatics. It includes 1,495,000 protein multiple sequence alignments (MSA) and an equal number of DNA MSAs for training, validation, and testing. These MSAs are simulated on randomly generated phylogenetic tree topologies, with branch lengths drawn from a uniform distribution, and sequences generated using the SpartaABC tool with parameters including insertion and deletion rates and Zipfian distribution parameters.
提供机构:
dotan1111
原始信息汇总
多序列比对作为序列到序列学习问题
数据
我们使用SpartaABC(Loewenthal et al., 2021)生成数百万个真实比对。SpartaABC需要以下输入:
- 一个有根的系统发育树,包括拓扑结构和分支长度;
- 替换模型(氨基酸或核苷酸);
- 根序列长度;
- 插入删除模型参数,包括:插入率(R_I),删除率(R_D),插入Zipfian分布参数(A_I),和删除Zipfian分布参数(A_D)。
通过使用ETE版本3.0(Huerta-Cepas et al., 2016)程序生成随机系统发育树拓扑结构,并使用默认参数模拟MSA。
我们生成了1,495,000、2,000和3,000个蛋白质MSA,分别用于训练、验证和测试数据。我们生成了相同数量的DNA MSA。对于每个随机树,分支长度从范围*(0.5,1.0)内的均匀分布中抽取。接下来,使用SpartaABC生成序列,参数如下:R_I,R_D in (0.0,0.05),A_I, A_D in (1.01,2.0)。比对长度以及树叶序列长度在数据集内部和之间变化,因为它们取决于插入删除动态和根长度。根长度在范围[32,44]*内均匀采样。除非另有说明,所有蛋白质数据集均使用WAG+G模型生成,所有DNA数据集均使用GTR+G模型生成,参数如下:
- 不同核苷酸的频率*(0.37, 0.166, 0.307, 0.158)*,顺序为"T", "C", "A"和"G";
- 替换率*(0.444, 0.0843, 0.116, 0.107, 0.00027)*,顺序为"a", "b", "c", "d", 和"e"。
示例
以下示例对应于上图中的MSA:
json {"MSA": "AAAC-GGG", "unaligned_seqs": {"seq0": "AAG", "seq1": "ACGG"}}


