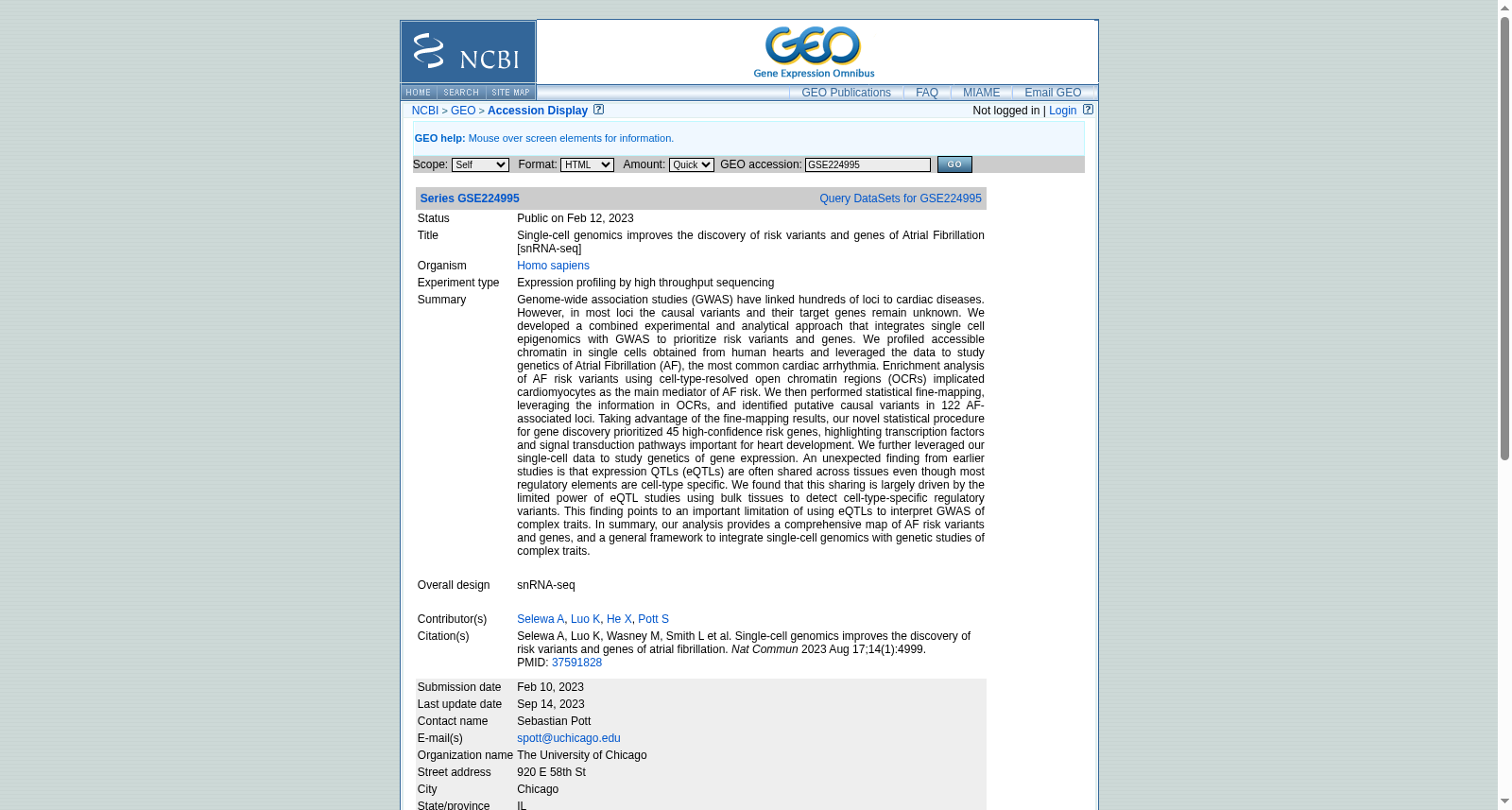
Single-cell genomics improves the discovery of risk variants and genes of Atrial Fibrillation [snRNA-seq]
收藏NIAID Data Ecosystem2026-05-01 收录
下载链接:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224995
下载链接
链接失效反馈官方服务:
资源简介:
Genome-wide association studies (GWAS) have linked hundreds of loci to cardiac diseases. However, in most loci the causal variants and their target genes remain unknown. We developed a combined experimental and analytical approach that integrates single cell epigenomics with GWAS to prioritize risk variants and genes. We profiled accessible chromatin in single cells obtained from human hearts and leveraged the data to study genetics of Atrial Fibrillation (AF), the most common cardiac arrhythmia. Enrichment analysis of AF risk variants using cell-type-resolved open chromatin regions (OCRs) implicated cardiomyocytes as the main mediator of AF risk. We then performed statistical fine-mapping, leveraging the information in OCRs, and identified putative causal variants in 122 AF-associated loci. Taking advantage of the fine-mapping results, our novel statistical procedure for gene discovery prioritized 45 high-confidence risk genes, highlighting transcription factors and signal transduction pathways important for heart development. We further leveraged our single-cell data to study genetics of gene expression. An unexpected finding from earlier studies is that expression QTLs (eQTLs) are often shared across tissues even though most regulatory elements are cell-type specific. We found that this sharing is largely driven by the limited power of eQTL studies using bulk tissues to detect cell-type-specific regulatory variants. This finding points to an important limitation of using eQTLs to interpret GWAS of complex traits. In summary, our analysis provides a comprehensive map of AF risk variants and genes, and a general framework to integrate single-cell genomics with genetic studies of complex traits. snRNA-seq
全基因组关联研究(Genome-wide association studies, GWAS)已将数百个基因座与心脏疾病建立关联。然而,绝大多数基因座中的致病变异及其靶基因仍未明确。本研究开发了一种实验与分析相结合的方法,将单细胞表观基因组学与GWAS整合,以优先筛选风险变异与靶基因。我们对提取自人类心脏的单细胞开放染色质开展了谱型分析,并依托该数据研究了心房颤动(Atrial Fibrillation, AF)——最常见的心脏心律失常——的遗传机制。通过细胞类型解析的开放染色质区域(OCRs)对AF风险变异进行富集分析,结果显示心肌细胞是AF风险的主要介导因子。随后,我们利用开放染色质区域的信息开展统计精细定位,在122个AF相关基因座中鉴定出潜在致病变异。借助精细定位结果,我们开发的新型基因发现统计流程优先筛选出45个高置信度风险基因,突出了对心脏发育至关重要的转录因子与信号转导通路。我们进一步利用单细胞数据研究了基因表达的遗传调控机制。此前研究有一项意外发现:尽管大多数调控元件具有细胞类型特异性,但表达数量性状基因座(expression QTLs, eQTLs)往往在多个组织中共享。我们发现,这种共享现象主要源于批量组织eQTL研究检测细胞类型特异性调控变异的能力有限。该发现指出了利用eQTLs解读复杂性状GWAS结果的一项重要局限。综上,本研究构建了AF风险变异与靶基因的全景图谱,并提供了一个将单细胞基因组学与复杂性状遗传研究相整合的通用框架。单细胞核RNA测序(snRNA-seq)
创建时间:
2023-09-14
搜集汇总
数据集介绍
背景与挑战
背景概述
该数据集利用单细胞核RNA测序技术,从3名捐赠者的12个心脏部位样本中,整合单细胞表观基因组学与GWAS数据,旨在识别心房颤动的风险变异和高置信度风险基因,重点揭示了心肌细胞在疾病风险中的关键作用,并为复杂性状的遗传研究提供了方法学框架。
以上内容由遇见数据集搜集并总结生成

暂无相关数据集


