taxonomy_phylum
收藏Hugging Face2025-07-25 更新2025-07-26 收录
下载链接:
https://huggingface.co/datasets/GleghornLab/taxonomy_phylum
下载链接
链接失效反馈官方服务:
资源简介:
这是一个基于UniProt数据库中经过审核的Swiss-Prot条目的数据集。数据集包含了条目名称、分类信息(界、门、纲、目、科、属、种)、序列信息以及基于门分类的标签。数据集经过了一系列预处理步骤,包括序列长度筛选、提取分类信息、去除缺失值、使用CD-HIT算法进行序列聚类、基于门分类创建标签、移除样本数量少于100的类别,并进行了分层划分,以保证训练集、验证集和测试集的样本分布。数据集分为训练集、验证集和测试集,分别包含258,989、5,000和5,000个样本。
提供机构:
Gleghorn Lab
创建时间:
2025-07-25
原始信息汇总
数据集概述
基本信息
- 数据集名称: GleghornLab/taxonomy_phylum
- 下载大小: 105,260,120 字节
- 数据集大小: 109,173,906 字节
数据特征
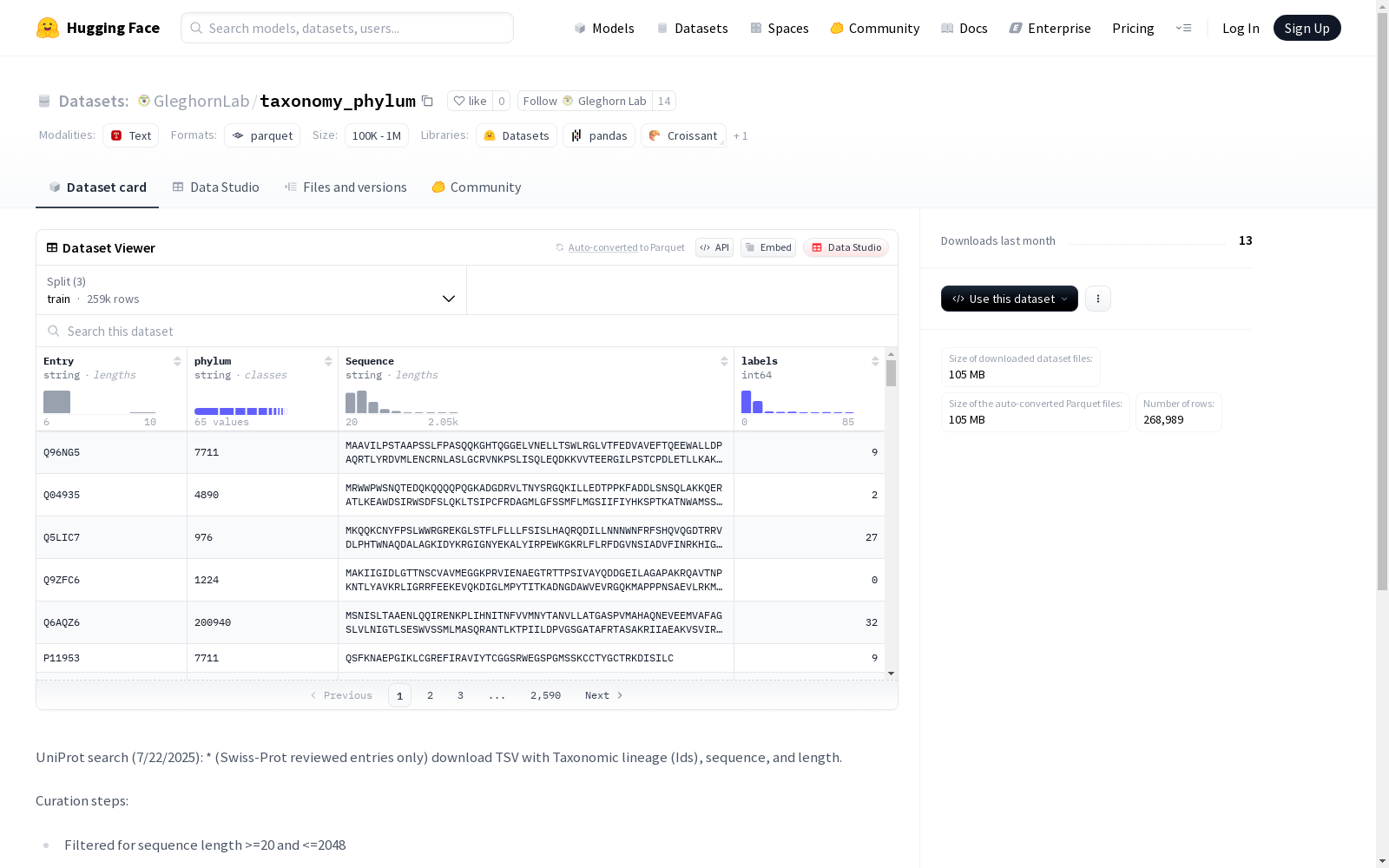
- 特征列:
Entry: 字符串类型phylum: 字符串类型Sequence: 字符串类型labels: 整数类型 (int64)
数据划分
- 训练集 (train):
- 样本数量: 258,989
- 大小: 105,120,975 字节
- 验证集 (valid):
- 样本数量: 5,000
- 大小: 2,034,095 字节
- 测试集 (test):
- 样本数量: 5,000
- 大小: 2,018,836 字节
数据来源与处理
- 数据来源: UniProt (Swiss-Prot reviewed entries only)
- 数据下载日期: 2025年7月22日
- 数据格式: TSV (包含分类学谱系ID、序列和长度)
数据预处理步骤
- 过滤序列长度在20到2048之间的条目。
- 从
taxonomic_lineage_ids列提取分类学ID(域、界、门、纲、目、科、属、种)。 - 保留
entry、phylum和sequence信息。 - 删除缺失值。
- 使用CD-HIT (Docker) 在80%相似度阈值和n=5的条件下进行聚类。
- 保留代表性序列。
- 根据
phylum创建标签。 - 删除样本数量少于100的
phylum/样本。 - 分层划分数据集:先划分测试集(5,000样本),再划分验证集(5,000样本),其余为训练集。
搜集汇总
数据集介绍
构建方式
在生物信息学领域,taxonomy_phylum数据集的构建体现了严谨的分子序列筛选流程。研究团队从UniProt数据库中精选Swiss-Prot已审核的蛋白质条目,通过TSV格式下载包含分类谱系标识、序列及长度的原始数据。采用多重过滤标准:限定序列长度在20至2048个氨基酸之间,运用CD-HIT工具以80%相似度阈值进行去冗余处理,保留代表性序列。为确保数据质量,剔除样本量不足100的门类,并采用分层抽样策略划分训练集、验证集和测试集。
特点
该数据集以其精细的生物分类标注著称,涵盖258,989条蛋白质序列及其对应的门级分类标签。每条记录包含UniProt登录号、序列字符串和数字化标签,序列长度分布经过严格把控。特别值得注意的是,数据经过去冗余处理并保持类别平衡,测试集与验证集各含5,000条样本,其余均纳入训练集,这种设计为分类模型的稳健评估提供了理想条件。
使用方法
研究者可利用该数据集开展蛋白质门级分类任务的基准测试,三个预划分的数据子集支持端到端的机器学习流程。输入特征为氨基酸序列字符串,输出标签对应35个细菌门类的分类编号。典型应用场景包括:构建序列特征提取器训练分类模型,评估跨域泛化能力,或作为预训练任务的生物学知识来源。数据以标准表格形式存储,可直接加载至主流深度学习框架进行建模。
背景与挑战
背景概述
taxonomy_phylum数据集是基于UniProt数据库中的Swiss-Prot条目构建的生物信息学资源,专注于生物分类学中的门(phylum)级别分类。该数据集由专业研究团队于2025年7月22日创建,通过严格的筛选和标注流程,整合了258,989条蛋白质序列及其对应的门级分类信息。其核心研究目标在于为生物序列分类提供高质量标注数据,推动计算生物学和机器学习在生物分类领域的应用。该数据集的构建采用了CD-HIT算法进行序列去冗余,并保持80%的相似度阈值,确保了数据的代表性和多样性。
当前挑战
taxonomy_phylum数据集面临的主要挑战体现在两个方面:在领域问题层面,生物序列的门级分类需要处理高度相似的序列特征和复杂的进化关系,这对机器学习模型的判别能力提出了极高要求;在构建过程层面,数据预处理涉及大规模序列去冗余(CD-HIT)、样本均衡化(删除少于100样本的门类)以及分层抽样等复杂操作,如何保持分类体系的完整性和数据分布的合理性成为关键难题。此外,原始数据中存在的标注噪声和序列长度变异(限制在20-2048个氨基酸之间)也增加了数据清洗的复杂度。
常用场景
经典使用场景
在生物信息学领域,taxonomy_phylum数据集为研究者提供了一个经过严格筛选的蛋白质序列分类基准。该数据集通过提取UniProt数据库中经过人工审核的Swiss-Prot条目,保留了具有代表性的蛋白质序列及其对应的门级分类信息。研究者可利用该数据集进行门级分类模型的训练与评估,探索不同蛋白质序列与生物分类学特征之间的关联。
实际应用
该数据集在生物医学研究中具有广泛用途,包括基因组注释辅助、新型蛋白质功能预测以及微生物群落分析。制药企业可利用该数据集训练的模型快速筛选具有特定功能的蛋白质,而环境科学家则能更准确地识别样本中的微生物组成。其标准化格式也便于整合到各类生物信息分析流程中。
衍生相关工作
基于taxonomy_phylum数据集,研究者已开发出多种深度学习方法用于蛋白质分类。典型工作包括结合Transformer架构的序列特征提取器,以及融合进化信息的图神经网络模型。这些衍生研究不仅提升了分类精度,还推动了蛋白质表示学习领域的发展,为后续的跨领域应用奠定了基础。
以上内容由遇见数据集搜集并总结生成


