HuggingFaceBio/clinvar-vep
收藏Hugging Face2026-06-18 更新2026-06-14 收录
下载链接:
https://hf-mirror.com/datasets/HuggingFaceBio/clinvar-vep
下载链接
链接失效反馈官方服务:
资源简介:
ClinVar VEP是一个用于评估DNA语言模型在临床变异致病性上的基准数据集,包含两个子集:coding(39,473个变异)和non-coding(15,258个变异)。每个子集都是一个平衡的二元分类任务,标签1表示致病/可能致病,标签0表示良性/可能良性。coding子集覆盖外显子蛋白编码区域,non-coding子集覆盖内含子及5′/3′非翻译区域(UTR)。数据集基于ClinVar变异数据构建,旨在为基因组学研究提供标准化评估工具。
A ClinVar variant-effect-prediction (VEP) benchmark, for evaluating DNA language models on clinical variant pathogenicity, with two subsets: coding (39,473 variants) and non-coding (15,258 variants). Each split is a balanced binary classification task: label = 1 for pathogenic / likely pathogenic and label = 0 for benign / likely benign. The coding subset covers exonic protein-coding regions, while the non-coding subset covers intronic and 5′/3′ UTR regions. The dataset is constructed from ClinVar variant data and serves as a standardized evaluation tool for genomics research.
提供机构:
HuggingFaceBio搜集汇总
数据集介绍
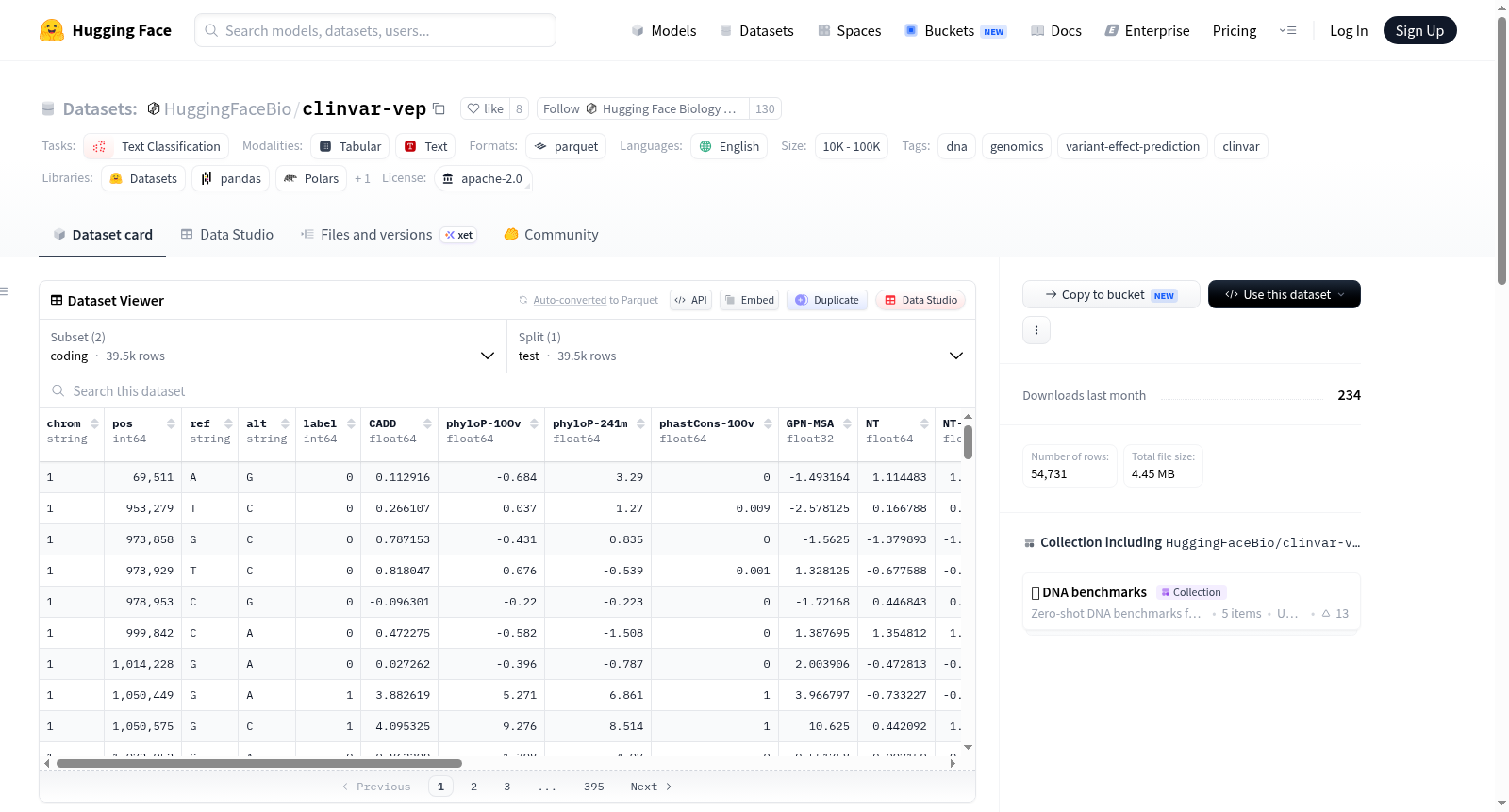
构建方式
该数据集基于ClinVar数据库,精心构建了一个用于评估DNA语言模型对临床变异致病性预测能力的基准测试。编码子集源自GPN-MSA研究中的`variant-effect-prediction`数据集,直接抽取其中39,473个编码区变异。非编码子集则通过严格流程独立构建:从原始ClinVar VCF文件中提取1-22号、X及Y染色体上的单核苷酸变异,依据临床标签将良性/可能良性映射为`label = 0`,致病性/可能致病性映射为`label = 1`,保留经过审阅的变异,并利用ClinVar后果术语进行区域分类,最终筛选出15,258个平衡的非编码变异。
特点
该数据集分为编码与非编码两个子集,各自构成平衡的二分类任务。编码子集包含39,473个变异,其中良性17,231个,致病性22,242个;非编码子集包含15,258个变异,良性与致病性各7,629个,实现了完美的类别平衡。非编码子集覆盖内含子区10,310个变异及非翻译区(5'UTR 4,174个、3'UTR 774个)变异,确保了区域多样性。每个子集均以parquet格式存储,方便高效加载。
使用方法
用户可通过Hugging Face的`datasets`库便捷加载数据,指定配置名称即可获取对应子集,如`load_dataset("hf-carbon/clinvar-vep-final", "coding", split="test")`加载编码子集,`load_dataset("hf-carbon/clinvar-vep-final", "non_coding", split="test")`加载非编码子集。数据集直接以文本分类任务形式提供,每条样本包含变异序列及对应的二元致病性标签,适用于评估各类DNA语言模型(如GPN-MSA)的临床变异效应预测能力。
背景与挑战
背景概述
在基因组学与精准医学领域,准确预测DNA变异的致病性是理解人类遗传疾病机制及推动临床诊断的关键挑战。ClinVar作为权威的人类变异注释数据库,为变异致病性分类提供了丰富标注,然而现有基准评测常面临数据分布不均、非编码区域覆盖不足等问题。为此,由Hugging Face Carbon团队于2026年构建并发布的clinvar-vep数据集,旨在系统评估DNA语言模型对临床变异致病性的预测能力。该研究团队整合了来自GPN-MSA研究(Benegas等,2025)和原始ClinVar VCF,精心设计了编码区与非编码区两个平衡子集,分别包含39,473和15,258个单核苷酸变异。这一基准填补了非编码区域致病性预测评测的空白,推动了基因组语言模型从编码区向全基因组范围的评估拓展,对计算生物学和临床基因组学领域产生了重要影响。
当前挑战
该数据集所解决的领域核心挑战在于,现有变异致病性预测方法多聚焦于编码区,而大量位于内含子及非翻译区的非编码变异对疾病贡献不可忽视,然而数据库标注匮乏且样本高度不平衡,严重制约模型泛化能力。针对此问题,clinvar-vep构建了首个大规模、类别均衡的非编码变异基准,通过严格筛选仅保留临床审核变异、平衡致病/良性比例至1:1,并细分为内含子和UTR亚型,显著提升了非编码区评测的科学性。在构建过程中,团队需从原始ClinVar VCF中处理数百万条记录,克服了染色体范围限制、标签二值映射、变异区域精细化分类等技术挑战,并确保编码与非编码子集数据兼容且无冗余,最终形成可复现、可扩展的标准化评测框架。
常用场景
经典使用场景
ClinVar-VEP数据集专为评估DNA语言模型在临床变异致病性预测任务中的表现而设计,是基因组学与自然语言处理交叉领域的标杆性基准。该数据集包含编码区与非编码区两个精心构建的子集,分别涵盖39,473个和15,258个变异位点,每个子集均为平衡二分类任务,其中致病性或可能致病性变异标注为1,良性或可能良性变异标注为0。研究者通常利用该数据集对预训练的DNA语言模型(如GPN-MSA、Carbon等)进行微调或零样本评估,以检验模型捕获序列上下文依赖致病性信号的能力。此外,该数据集还可用于比较不同模型架构(如Transformer与卷积神经网络)在基因组区域特异性上的表现差异。
实际应用
在精准医学与临床诊断实践中,该数据集可支撑自动化变异解读系统的开发与验证。临床实验室可借助基于ClinVar-VEP训练的预测模型,对全基因组测序中识别的新发或罕见变异进行快速致病性分级,辅助遗传咨询中的基因检测报告生成。药物基因组学领域亦能受益于此,通过评估非编码区调控变异对药物代谢酶或靶点基因表达的影响,为个体化用药方案的制定提供基因组层面的证据。此外,该基准还可用于优化临床测序数据质量控制流程,减少良性变异被误判为致病性所带来的不必要诊断焦虑与医疗资源消耗。
衍生相关工作
该数据集作为基因组语言模型评估基准,已衍生出多项关键研究。原始构建工作发表于Carbon项目(Allal et al., 2026),其中利用平衡非编码子集证明了DNA语言模型在非编码区致病性预测上的有效性。此前,GPN-MSA研究(Benegas et al., 2025)曾使用其编码子集评估多序列比对增强的变异效应预测性能。此外,该基准被广泛用于对比不同基因组预训练策略(如掩码语言建模与对比学习)在下游临床任务中的迁移表现,并催生了针对变体偏好去偏、数据泄露检测以及长序列上下文建模等方法的优化工作,推动了基因组基础模型在可重复性与公平性评估方面的标准化进程。
以上内容由遇见数据集搜集并总结生成


