SABmark-dataset
收藏Hugging Face2025-09-16 更新2025-09-17 收录
下载链接:
https://huggingface.co/datasets/DeepFoldProtein/SABmark-dataset
下载链接
链接失效反馈官方服务:
资源简介:
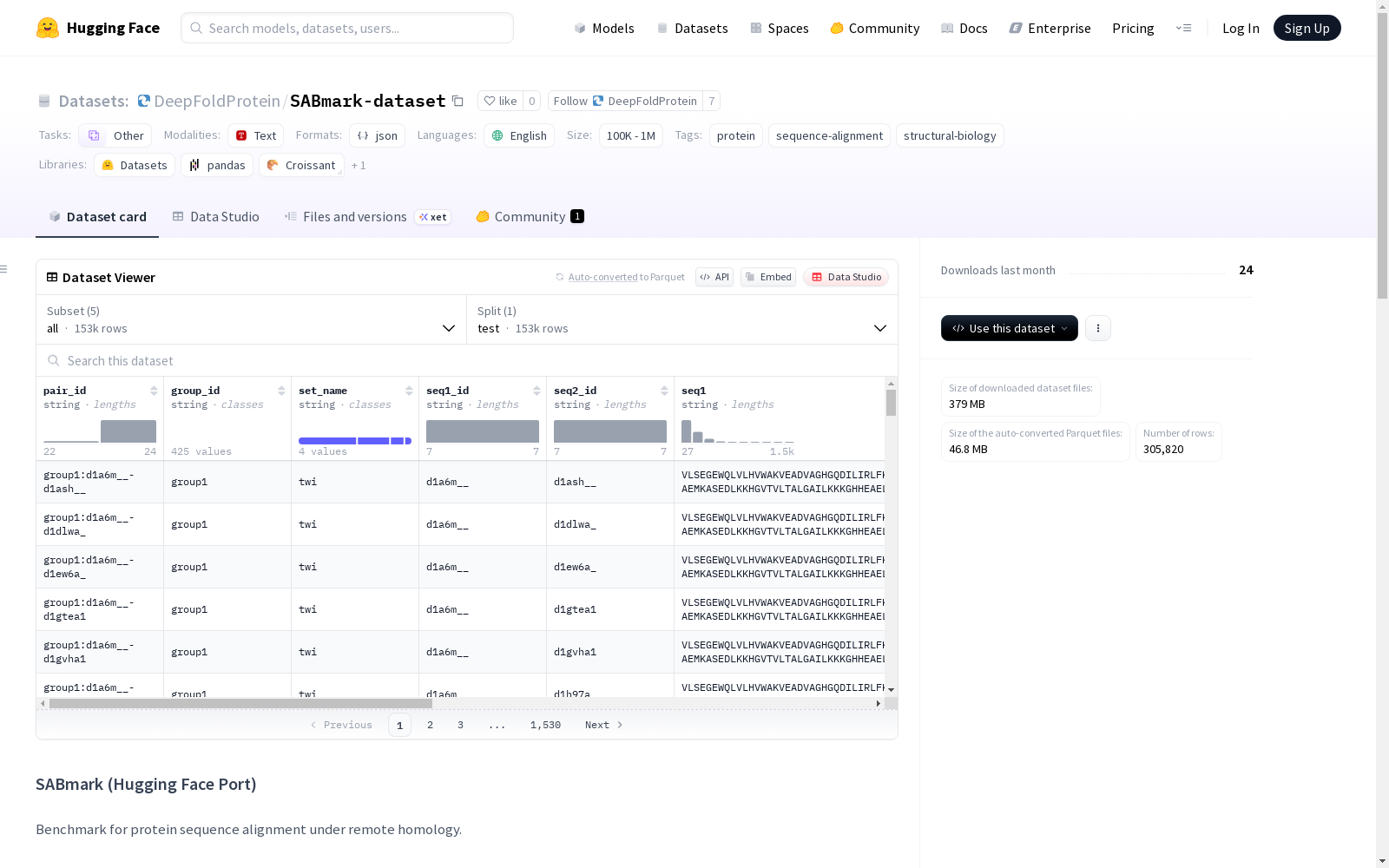
SABmark是一个蛋白质序列对齐的基准数据集,用于远程同源检测。它包括几个子集:twi(<25%相似度),sup(低到中等相似度),twi_fp和sup_fp(分别添加了虚假阳性的twi和sup子集)。数据集的特征包括序列对ID、组ID、序列名称、参考对齐列表、序列身份百分比、SCOP标签和元数据。
SABmark is a benchmark dataset for protein sequence alignment, designed for remote homology detection. It includes several subsets: twi (<25% sequence identity), sup (low to moderate sequence identity), twi_fp and sup_fp (the twi and sup subsets with false positives added, respectively). The features of this dataset consist of sequence pair ID, group ID, sequence name, reference alignment list, percentage sequence identity, SCOP label, and metadata.
提供机构:
DeepFoldProtein创建时间:
2025-09-16
原始信息汇总
SABmark数据集概述
数据集简介
SABmark是一个用于远程同源性蛋白质序列比对的基准数据集,涵盖整个已知折叠空间。
子集配置
数据集包含5个子集配置:
- twi:Twilight子集(<25%序列一致性)
- sup:超家族子集(低到中等一致性)
- twi_fp:Twilight子集(添加假阳性样本)
- sup_fp:超家族子集(添加假阳性样本)
- all:所有子集的并集
数据特征
每个样本包含以下特征字段:
- 标识信息:
pair_id、group_id、set_name - 序列信息:
seq1_id、seq2_id、seq1、seq2 - 比对信息:
ref_alignment(基于0的索引对列表) - 统计信息:
percent_identity、scop_labels、meta
数据格式
- 数据文件格式:JSON Lines (.jsonl)
- 所有子集仅包含测试集分割
使用方式
python from datasets import load_dataset ds = load_dataset("DeepFoldProtein/SABmark", name="twi", split="test") ex = ds[0] print(ex["seq1"], ex["seq2"], ex["ref_alignment"][:5])
引用信息
bibtex @article{VanWalle2004SABmark, title={SABmark---a benchmark for sequence alignment that covers the entire known fold space}, author={Van Walle, Ivan and Lasters, Ignace and Wyns, Lode}, journal={Bioinformatics}, volume={21}, number={7}, pages={1267--1268}, year={2004}, publisher={Oxford University Press}, DOI = {10.1093/bioinformatics/bth493} }
搜集汇总
数据集介绍
构建方式
在结构生物学领域,SABmark数据集通过精心筛选蛋白质序列对构建而成,涵盖远缘同源关系下的序列比对场景。其构建过程基于SCOP数据库的结构分类,特别设计了黄昏子集(序列一致性低于25%)和超家族子集,并引入包含错误比对的扩展版本以增强评估鲁棒性。所有数据均以标准化JSONL格式存储,确保序列信息和参考比对结果的完整性。
特点
该数据集的核心特征在于全面覆盖已知折叠空间的蛋白质远缘同源关系,提供精确的序列一致性百分比和SCOP分类标签。每个样本包含配对的蛋白质序列标识符、原始氨基酸序列以及基于结构比对的黄金标准参考对齐位置。特别设计的错误阳性子集为评估比对算法的特异性提供了独特价值,而丰富的元数据字段支持多维度分析。
使用方法
研究人员可通过HuggingFace数据集库直接加载指定子集,例如使用load_dataset函数调用'twi'配置获取黄昏子集。加载后的数据对象包含完整的序列对信息和参考对齐索引,支持直接计算比对精度指标。典型应用包括蛋白质序列比对算法验证、远缘同源检测模型训练以及结构生物信息学方法的基准测试。
背景与挑战
背景概述
蛋白质序列比对作为结构生物学的基础分析方法,其准确性直接关系到蛋白质功能预测与进化关系研究的可靠性。SABmark数据集由Van Walle等人于2004年创建,旨在构建覆盖已知折叠空间的基准测试集,通过系统性地整合不同同源性层级的蛋白质序列对,为远程同源性检测算法提供标准化评估框架。该数据集通过科学分类体系将蛋白质序列划分为超家族(superfamilies)和暮光区(twilight)子集,显著推动了蛋白质序列比对算法在低相似度场景下的性能优化,成为计算生物学领域的重要基准资源。
当前挑战
该数据集核心挑战在于解决远程同源性检测中序列相似性低于25%的蛋白质对齐问题,此类序列虽具有结构相似性却难以通过传统序列比对方法识别。构建过程中需克服蛋白质折叠空间全面覆盖的技术难题,包括从SCOP数据库提取结构域、严格过滤序列冗余性,以及人工验证参考比对的准确性。此外,为增强算法鲁棒性,数据集专门引入负样本构建模块,通过插入错误配对序列来检验算法对假阳性结果的判别能力,这一设计对数据标注的精确性和生物学合理性提出了极高要求。
常用场景
经典使用场景
在结构生物学领域,SABmark数据集作为蛋白质序列比对的金标准基准测试集,主要应用于评估远程同源关系下的序列比对算法性能。其经典使用场景涵盖了对不同同源层级(如超家族和暮光区)的蛋白质序列对进行比对精度测试,特别是在序列一致性低于25%的极端条件下验证算法的鲁棒性。研究人员通过该数据集能够系统性地检验比对模型在进化距离较远的蛋白质序列上的表现,为算法优化提供关键依据。
实际应用
在实际应用层面,SABmark数据集被广泛应用于蛋白质结构预测、功能注释和药物设计等领域。制药公司利用该数据集训练的比对算法来识别潜在药物靶点与已知蛋白质的进化关系,加速新药发现进程。生物信息学工具开发者则依赖其评估软件在真实生物学场景中的实用性,确保比对结果能够可靠支撑下游的蛋白质功能分析和结构建模工作。这些应用直接促进了精准医疗和蛋白质工程设计的发展。
衍生相关工作
基于SABmark数据集衍生出了大量经典研究工作,包括深度学习方法在蛋白质比对中的应用突破。许多神经网络架构如循环神经网络和注意力机制模型都使用该数据集进行性能验证,推动了DeepFam、ProtTrans等先进模型的诞生。这些工作不仅扩展了计算生物学的研究边界,还催生了新的交叉学科方向,如基于深度学习的蛋白质工程和进化分析,为整个领域带来了方法论上的革新。
以上内容由遇见数据集搜集并总结生成


