Trelis/protein_stability_single_mutation
收藏Hugging Face2023-08-21 更新2024-03-04 收录
下载链接:
https://hf-mirror.com/datasets/Trelis/protein_stability_single_mutation
下载链接
链接失效反馈官方服务:
资源简介:
---
task_categories:
- question-answering
- tabular-classification
- text-generation
language:
- en
tags:
- biology
- proteins
- amino-acids
size_categories:
- 100K<1M
---
# Protein Data Stability - Single Mutation
This repository contains data on the change in protein stability with a single mutation.
## Attribution of Data Sources
- **Primary Source**: Tsuboyama, K., Dauparas, J., Chen, J. et al. Mega-scale experimental analysis of protein folding stability in biology and design. Nature 620, 434–444 (2023). [Link to the paper](https://www.nature.com/articles/s41586-023-06328-6)
- **Dataset Link**: [Zenodo Record](https://zenodo.org/record/7992926)
As to where the dataset comes from in this broader work, the relevant dataset (#3) is shown in `dataset_table.jpeg` of this repository's files.
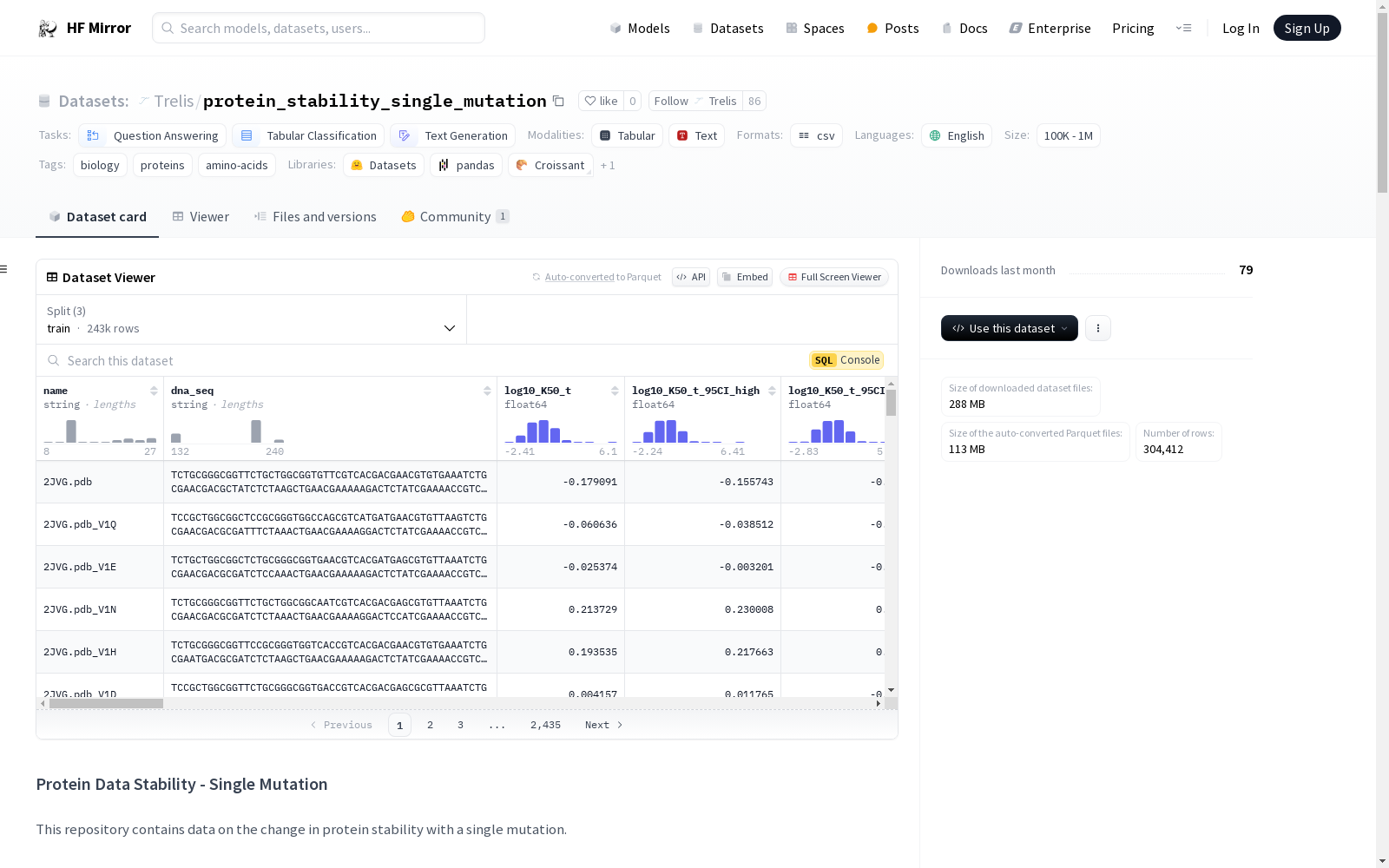
## Sample Protein Stability Data [subset of 4 columns]
| Base Protein Sequence | Mutation | ΔΔG_ML | Classification |
|-------------------------------------------------------------|----------|--------------------|-----------------|
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63W | -0.2010871345320799 | neutral |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63Y | 0.0194756159891467 | neutral |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63F | 0.7231614929744659 | stabilising |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63P | -0.3668887752897785 | neutral |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63C | -0.5317304030261774 | destabilising |
## Dataset Structure
This dataset focuses on the differential deltaG of *unfolding* (mutation minus base) of various protein mutations and is derived from stability measurements (free energy of unfolding) measured by two proteases, trypsin and chymotrypsin.
### Columns (Trypsin):
- **name**: The name of the protein variant.
- **dna_seq**: The DNA sequence encoding the protein variant.
- **log10_K50_t**: The log10 of the K50 value measured with trypsin (a measure of stability).
- **log10_K50_t_95CI_high**: The upper bound of the 95% confidence interval for log10_K50_t.
- **log10_K50_t_95CI_low**: The lower bound of the 95% confidence interval for log10_K50_t.
- **log10_K50_t_95CI**: The width of the 95% confidence interval for log10_K50_t.
- **fitting_error_t**: A measure of error between the model and data for trypsin.
- **log10_K50unfolded_t**: The predicted log10 K50 value for the unfolded state with trypsin.
- **deltaG_t**: The ΔG stability calculated from the trypsin data.
- **deltaG_t_95CI_high**: The upper bound of the ΔG confidence interval from trypsin.
- **deltaG_t_95CI_low**: The lower bound of the ΔG confidence interval from trypsin.
- **deltaG_t_95CI**: The width of the ΔG confidence interval from trypsin.
### Columns (Chymotrypsin):
- **log10_K50_c**: Analogous to `log10_K50_t`, but for chymotrypsin.
- **log10_K50_c_95CI_high**: Upper bound of the 95% CI for `log10_K50_c`.
- **log10_K50_c_95CI_low**: Lower bound of the 95% CI for `log10_K50_c`.
- **log10_K50_c_95CI**: Width of the 95% CI for `log10_K50_c`.
- **fitting_error_c**: A measure of error between the model and data for chymotrypsin.
- **log10_K50unfolded_c**: Predicted log10 K50 value for the unfolded state with chymotrypsin.
- **deltaG_c**: ΔG stability calculated from the chymotrypsin data.
- **deltaG_c_95CI_high**: Upper bound of the ΔG CI from chymotrypsin.
- **deltaG_c_95CI_low**: Lower bound of the ΔG CI from chymotrypsin.
- **deltaG_c_95CI**: Width of the ΔG CI from chymotrypsin.
### Combined Data:
- **deltaG**: The combined ΔG estimate from both trypsin and chymotrypsin.
- **deltaG_95CI_high**: Upper bound of the combined ΔG confidence interval.
- **deltaG_95CI_low**: Lower bound of the combined ΔG confidence interval.
- **deltaG_95CI**: Width of the combined ΔG confidence interval.
### Protein Sequencing Data:
- **aa_seq_full**: The full amino acid sequence.
- **aa_seq**: A (sometimes shortened) amino acid sequence representing the protein.
- **mut_type**: The type of mutation introduced to the protein.
- **WT_name**: Name of the wild type variant.
- **WT_cluster**: Cluster classification for the wild type variant.
- **mutation**: Represented as a combination of amino acid and its position (e.g., F10N indicates changing the 10th amino acid (F) in a sequence for N).
- **base_aa_seq**: The base sequence of the protein before the mutation.
### Derived Data:
- **log10_K50_trypsin_ML**: Log10 value of K50 derived from a machine learning model using trypsin data.
- **log10_K50_chymotrypsin_ML**: Log10 value of K50 derived from a machine learning model using chymotrypsin data.
- **dG_ML**: ΔG derived from a machine learning model that makes use of stability measurements from both proteases.
- **ddG_ML**: Differential ΔG (mutation minus base) derived from a machine learning model.
### Classification:
- **Stabilizing_mut**: Indicates whether the mutation is stabilizing or not.
- **pair_name**: Name representation combining the wild type and mutation.
- **classification**: Classification based on `ddG_ML`:
- Rows below -0.5 standard deviations are classified as 'destabilising'.
- Rows above +0.5 standard deviations are classified as 'stabilising'.
- Rows between -0.5 and 0.5 standard deviations are classified as 'neutral'.
This dataset offers a comprehensive view of protein mutations, their effects, and how they relate to the stability measurements made with trypsin and chymotrypsin.
### Understanding ΔG (delta G)
ΔG is the Gibbs free energy change of a process, dictating whether a process is thermodynamically favorable:
- **Negative ΔG**: Indicates the process is energetically favorable. For protein unfolding, it implies the protein is more stable in its unfolded form.
- **Positive ΔG**: Indicates the process is not energetically favorable. In protein unfolding, it means the protein requires energy to maintain its unfolded state, i.e. it is stable in folded form.
The **delta delta G** (ΔΔG) represents the deltaG of the mutation compared to the base protein:
- **Positive ΔΔG**: Suggests the mutation enhances protein stability.
- **Negative ΔΔG**: Suggests the mutation decreases protein stability.
### Data Cleanup and Validation:
1. Filtering: The dataset has been curated to only include examples of single mutations.
2. Sequence mutations were extracted from the row names. Base mutations are labelled as 'base'.
3. Consistency Check: Only rows with a consistent 'mutation', aligned with both the base and mutated sequences from the raw data, have been retained.
任务类别:
- 问答
- 表格分类
- 文本生成
语言:英语
标签:
- 生物学
- 蛋白质
- 氨基酸
数据规模:10万至100万条
# 蛋白质数据稳定性——单突变
本仓库收录了单突变引发的蛋白质稳定性变化相关数据。
## 数据来源标注
- **主要来源**:Tsuboyama, K., Dauparas, J., Chen, J. 等. 生物学与设计领域蛋白质折叠稳定性的大规模实验分析. 《自然》(Nature), 620, 434–444 (2023). [论文链接](https://www.nature.com/articles/s41586-023-06328-6)
- **数据集链接**:[Zenodo 存档记录](https://zenodo.org/record/7992926)
在这项更广泛的研究中,本仓库文件中的`dataset_table.jpeg`展示了相关的第3号数据集。
## 蛋白质稳定性数据示例(4列子集)
| 原始蛋白质序列 | 突变位点 | ΔΔG_ML | 分类 |
|-------------------------------------------------------------|----------|--------------------|-----------------|
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63W | -0.2010871345320799 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63Y | 0.0194756159891467 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63F | 0.7231614929744659 | 稳定化 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63P | -0.3668887752897785 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63C | -0.5317304030261774 | 去稳定化 |
## 数据集结构
本数据集聚焦于多种蛋白质突变的解折叠ΔΔG值(突变型与野生型的ΔG差值),其数据来源于两种蛋白酶——胰蛋白酶(trypsin)与胰凝乳蛋白酶(chymotrypsin)的稳定性测量结果(解折叠自由能)。
### 胰蛋白酶(trypsin)相关列:
- **name**:蛋白质变体的名称
- **dna_seq**:编码该蛋白质变体的DNA序列
- **log10_K50_t**:胰蛋白酶检测下测得的K50值的对数10转换值(稳定性衡量指标)
- **log10_K50_t_95CI_high**:log10_K50_t的95%置信区间上限
- **log10_K50_t_95CI_low**:log10_K50_t的95%置信区间下限
- **log10_K50_t_95CI**:log10_K50_t的95%置信区间宽度
- **fitting_error_t**:胰蛋白酶模型与实验数据的拟合误差衡量指标
- **log10_K50unfolded_t**:胰蛋白酶检测下蛋白质未折叠状态的预测log10 K50值
- **deltaG_t**:基于胰蛋白酶数据计算得到的ΔG稳定性值
- **deltaG_t_95CI_high**:胰蛋白酶检测得到的ΔG置信区间上限
- **deltaG_t_95CI_low**:胰蛋白酶检测得到的ΔG置信区间下限
- **deltaG_t_95CI**:胰蛋白酶检测得到的ΔG置信区间宽度
### 胰凝乳蛋白酶(chymotrypsin)相关列:
- **log10_K50_c**:与`log10_K50_t`定义一致,但对应胰凝乳蛋白酶检测的数据
- **log10_K50_c_95CI_high**:`log10_K50_c`的95%置信区间上限
- **log10_K50_c_95CI_low**:`log10_K50_c`的95%置信区间下限
- **log10_K50_c_95CI**:`log10_K50_c`的95%置信区间宽度
- **fitting_error_c**:胰凝乳蛋白酶模型与实验数据的拟合误差衡量指标
- **log10_K50unfolded_c**:胰凝乳蛋白酶检测下蛋白质未折叠状态的预测log10 K50值
- **deltaG_c**:基于胰凝乳蛋白酶数据计算得到的ΔG稳定性值
- **deltaG_c_95CI_high**:胰凝乳蛋白酶检测得到的ΔG置信区间上限
- **deltaG_c_95CI_low**:胰凝乳蛋白酶检测得到的ΔG置信区间下限
- **deltaG_c_95CI**:胰凝乳蛋白酶检测得到的ΔG置信区间宽度
### 合并数据:
- **deltaG**:结合胰蛋白酶与胰凝乳蛋白酶数据得到的综合ΔG估计值
- **deltaG_95CI_high**:综合ΔG的95%置信区间上限
- **deltaG_95CI_low**:综合ΔG的95%置信区间下限
- **deltaG_95CI**:综合ΔG的95%置信区间宽度
### 蛋白质测序数据:
- **aa_seq_full**:完整的氨基酸序列
- **aa_seq**:代表该蛋白质的(有时为缩短版)氨基酸序列
- **mut_type**:引入的蛋白质突变类型
- **WT_name**:野生型变体的名称
- **WT_cluster**:野生型变体的聚类分类
- **mutation**:以氨基酸与位点组合形式表示的突变(例如F10N代表将序列中第10位氨基酸F替换为N)
- **base_aa_seq**:突变前的蛋白质原始氨基酸序列
### 衍生数据:
- **log10_K50_trypsin_ML**:基于胰蛋白酶数据通过机器学习模型得到的K50值的对数10转换值
- **log10_K50_chymotrypsin_ML**:基于胰凝乳蛋白酶数据通过机器学习模型得到的K50值的对数10转换值
- **dG_ML**:结合两种蛋白酶的稳定性测量数据,通过机器学习模型得到的ΔG值
- **ddG_ML**:通过机器学习模型得到的ΔΔG值(突变型与野生型的ΔG差值)
### 分类标签:
- **Stabilizing_mut**:标记该突变是否具有稳定蛋白质的作用
- **pair_name**:结合野生型与突变信息的命名表示
- **classification**:基于`ddG_ML`的分类结果:
- 低于-0.5倍标准差的样本被归类为「去稳定化」
- 高于+0.5倍标准差的样本被归类为「稳定化」
- 介于-0.5至0.5倍标准差之间的样本被归类为「中性」
本数据集全面涵盖了蛋白质突变、其功能效应,以及与胰蛋白酶和胰凝乳蛋白酶稳定性测量结果的关联关系。
### ΔG(吉布斯自由能)释义:
ΔG是某一过程的吉布斯自由能变化量(Gibbs Free Energy Change),用于判断该过程是否具有热力学可行性:
- **ΔG为负值**:表明该过程在能量上是有利的。对于蛋白质解折叠过程而言,意味着蛋白质的未折叠形式更稳定。
- **ΔG为正值**:表明该过程在能量上不具备自发进行的条件。在蛋白质解折叠过程中,则表示蛋白质需要外界能量才能维持未折叠状态,即其折叠形式更稳定。
**ΔΔG(delta delta G)**代表突变型与野生型蛋白质之间的ΔG差值:
- **ΔΔG为正值**:提示该突变可增强蛋白质稳定性
- **ΔΔG为负值**:提示该突变会降低蛋白质稳定性
### 数据清理与验证:
1. 筛选流程:本数据集已经过整理,仅保留单突变样本
2. 突变提取:从行名称中提取序列突变信息,野生型样本标注为「base」
3. 一致性校验:仅保留突变信息与原始数据中的原始、突变序列均匹配的样本
提供机构:
Trelis
原始信息汇总
蛋白质数据稳定性 - 单点突变
该数据集包含单点突变对蛋白质稳定性变化的数据。
数据来源
- 主要来源: Tsuboyama, K., Dauparas, J., Chen, J. 等人. Mega-scale experimental analysis of protein folding stability in biology and design. Nature 620, 434–444 (2023).
- 数据集链接: Zenodo Record
样本蛋白质稳定性数据 [部分列]
| 基础蛋白质序列 | 突变 | ΔΔG_ML | 分类 |
|---|---|---|---|
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63W | -0.2010871345320799 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63Y | 0.0194756159891467 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63F | 0.7231614929744659 | 稳定化 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63P | -0.3668887752897785 | 中性 |
| FDIYVVTADYLPLGAEQDAITLREGQYVEVLDAAHPLRWLVRTKPTKSSPSRQGWVSPAYLDRRL | R63C | -0.5317304030261774 | 不稳定化 |
数据集结构
该数据集关注各种蛋白质突变的展开的差分deltaG(突变减去基础),并从稳定性测量(展开自由能)中得出,这些测量由两种蛋白酶——胰蛋白酶和糜蛋白酶进行。
列(胰蛋白酶):
- name: 蛋白质变体的名称。
- dna_seq: 编码蛋白质变体的DNA序列。
- log10_K50_t: 用胰蛋白酶测量的K50值的log10(稳定性的度量)。
- log10_K50_t_95CI_high: log10_K50_t的95%置信区间的上限。
- log10_K50_t_95CI_low: log10_K50_t的95%置信区间的下限。
- log10_K50_t_95CI: log10_K50_t的95%置信区间的宽度。
- fitting_error_t: 模型与胰蛋白酶数据之间的误差度量。
- log10_K50unfolded_t: 用胰蛋白酶预测的展开状态的log10 K50值。
- deltaG_t: 从胰蛋白酶数据计算的ΔG稳定性。
- deltaG_t_95CI_high: 胰蛋白酶的ΔG置信区间的上限。
- deltaG_t_95CI_low: 胰蛋白酶的ΔG置信区间的下限。
- deltaG_t_95CI: 胰蛋白酶的ΔG置信区间的宽度。
列(糜蛋白酶):
- log10_K50_c: 类似于
log10_K50_t,但用于糜蛋白酶。 - log10_K50_c_95CI_high:
log10_K50_c的95%置信区间的上限。 - log10_K50_c_95CI_low:
log10_K50_c的95%置信区间的下限。 - log10_K50_c_95CI:
log10_K50_c的95%置信区间的宽度。 - fitting_error_c: 模型与糜蛋白酶数据之间的误差度量。
- log10_K50unfolded_c: 用糜蛋白酶预测的展开状态的log10 K50值。
- deltaG_c: 从糜蛋白酶数据计算的ΔG稳定性。
- deltaG_c_95CI_high: 糜蛋白酶的ΔG置信区间的上限。
- deltaG_c_95CI_low: 糜蛋白酶的ΔG置信区间的下限。
- deltaG_c_95CI: 糜蛋白酶的ΔG置信区间的宽度。
合并数据:
- deltaG: 从胰蛋白酶和糜蛋白酶合并的ΔG估计。
- deltaG_95CI_high: 合并的ΔG置信区间的上限。
- deltaG_95CI_low: 合并的ΔG置信区间的下限。
- deltaG_95CI: 合并的ΔG置信区间的宽度。
蛋白质序列数据:
- aa_seq_full: 完整的氨基酸序列。
- aa_seq: 代表蛋白质的(有时缩短的)氨基酸序列。
- mut_type: 引入蛋白质的突变类型。
- WT_name: 野生型变体的名称。
- WT_cluster: 野生型变体的聚类分类。
- mutation: 表示为氨基酸及其位置的组合(例如,F10N表示将序列中第10个氨基酸(F)改为N)。
- base_aa_seq: 突变前的蛋白质基础序列。
衍生数据:
- log10_K50_trypsin_ML: 使用胰蛋白酶数据的机器学习模型衍生的K50的log10值。
- log10_K50_chymotrypsin_ML: 使用糜蛋白酶数据的机器学习模型衍生的K50的log10值。
- dG_ML: 使用两种蛋白酶的稳定性测量数据的机器学习模型衍生的ΔG。
- ddG_ML: 机器学习模型衍生的差分ΔG(突变减去基础)。
分类:
- Stabilizing_mut: 指示突变是否稳定。
- pair_name: 结合野生型和突变的名称表示。
- classification: 基于
ddG_ML的分类:- 低于-0.5标准差的行被分类为不稳定化。
- 高于+0.5标准差的行被分类为稳定化。
- 介于-0.5和0.5标准差之间的行被分类为中性。
该数据集提供了蛋白质突变、其影响以及它们与胰蛋白酶和糜蛋白酶稳定性测量的关系的全面视图。
理解ΔG(delta G)
ΔG是过程的吉布斯自由能变化,决定过程是否在热力学上有利:
- 负ΔG: 表示过程在能量上有利。对于蛋白质展开,这意味着蛋白质在其展开形式中更稳定。
- 正ΔG: 表示过程在能量上不利。对于蛋白质展开,这意味着蛋白质需要能量来维持其展开状态,即它在折叠形式中稳定。
差分ΔG(ΔΔG)表示突变与基础蛋白质的ΔG比较:
- 正ΔΔG: 表明突变增强了蛋白质稳定性。
- 负ΔΔG: 表明突变降低了蛋白质稳定性。
数据清理和验证:
- 过滤: 数据集已筛选,仅包括单点突变的示例。
- 序列突变从行名称中提取。基础突变标记为base。
- 一致性检查: 仅保留与原始数据中的基础和突变序列一致的mutation行。
搜集汇总
数据集介绍
构建方式
该数据集聚焦于蛋白质单点突变对稳定性的影响,通过实验测量蛋白质的自由能变化(ΔG)来构建。数据来源于Tsuboyama等人在2023年发表的研究,涵盖了从两种蛋白酶(胰蛋白酶和糜蛋白酶)中获得的稳定性测量值。数据集通过机器学习模型进一步推导出ΔG和ΔΔG值,并结合实验数据进行验证和清理,确保仅包含单点突变的数据,并保持突变与原始序列的一致性。
特点
该数据集的显著特点在于其全面性和精确性,涵盖了蛋白质序列、突变类型、稳定性测量值及其置信区间等多维度信息。通过结合胰蛋白酶和糜蛋白酶的测量数据,数据集提供了对蛋白质突变稳定性变化的深入理解。此外,数据集还通过机器学习模型生成了ΔG和ΔΔG的预测值,为研究蛋白质稳定性提供了多层次的分析工具。
使用方法
该数据集适用于多种生物信息学任务,包括蛋白质稳定性预测、突变效应分类和文本生成等。用户可以通过分析ΔΔG值来评估突变对蛋白质稳定性的影响,并利用分类信息(如稳定、中性或不稳定)进行进一步的生物学研究。数据集的结构化格式和详细的元数据使得其在机器学习模型训练和验证中具有广泛的应用潜力。
背景与挑战
背景概述
蛋白质稳定性研究在生物学和生物工程领域具有重要意义,尤其是在理解蛋白质折叠与突变对其稳定性的影响方面。Trelis/protein_stability_single_mutation数据集由Tsuboyama等人于2023年创建,基于大规模实验分析蛋白质折叠稳定性的研究成果。该数据集的核心研究问题在于通过单点突变评估蛋白质稳定性的变化,并提供了详细的实验数据,涵盖了多种蛋白质突变及其对应的ΔΔG值。这一数据集不仅为蛋白质工程和设计提供了宝贵的实验依据,还为机器学习模型在蛋白质稳定性预测中的应用奠定了基础。
当前挑战
该数据集在构建过程中面临多项挑战。首先,实验数据的获取与处理需要高精度的测量技术,尤其是在使用胰蛋白酶和糜蛋白酶进行稳定性测定时,确保数据的准确性和一致性至关重要。其次,数据集的分类和标注依赖于复杂的生物学知识,如何准确地将突变分类为稳定、中性或不稳定突变,是一个具有挑战性的任务。此外,数据集的规模和多样性要求高效的算法和模型来处理和分析,以提取有用的生物学信息。最后,确保数据集的透明性和可重复性,以便其他研究者能够验证和扩展这些发现,也是一项重要的挑战。
常用场景
经典使用场景
在蛋白质生物学领域,Trelis/protein_stability_single_mutation数据集的经典应用场景主要集中在蛋白质稳定性预测与突变效应分析。该数据集通过记录单点突变对蛋白质稳定性的影响,为研究人员提供了一个详尽的突变效应数据库。通过分析ΔΔG值,研究者能够预测特定突变对蛋白质折叠稳定性的影响,从而为蛋白质工程和药物设计提供理论依据。
衍生相关工作
基于Trelis/protein_stability_single_mutation数据集,许多相关研究工作得以展开。例如,研究人员利用该数据集开发了预测蛋白质突变效应的机器学习模型,进一步提高了突变效应预测的准确性。此外,该数据集还被用于验证和优化蛋白质设计算法,推动了蛋白质工程领域的技术进步。这些衍生工作不仅丰富了蛋白质生物学的研究内容,也为实际应用提供了技术支持。
数据集最近研究
最新研究方向
在蛋白质生物学领域,Trelis/protein_stability_single_mutation数据集的最新研究方向主要集中在通过大规模实验分析蛋白质折叠稳定性的变化,特别是单点突变对蛋白质稳定性的影响。该数据集结合了机器学习模型与实验数据,提供了对蛋白质突变后稳定性变化的详细分类和量化分析,为蛋白质工程和药物设计提供了重要的理论支持。研究者们正利用这一数据集探索蛋白质突变与稳定性之间的复杂关系,以期在生物技术和医学领域取得突破性进展。
以上内容由遇见数据集搜集并总结生成


