Benchmark_Dataset-Human_population_classification
收藏Hugging Face2025-10-23 更新2025-10-24 收录
下载链接:
https://huggingface.co/datasets/BGI-HangzhouAI/Benchmark_Dataset-Human_population_classification
下载链接
链接失效反馈官方服务:
资源简介:
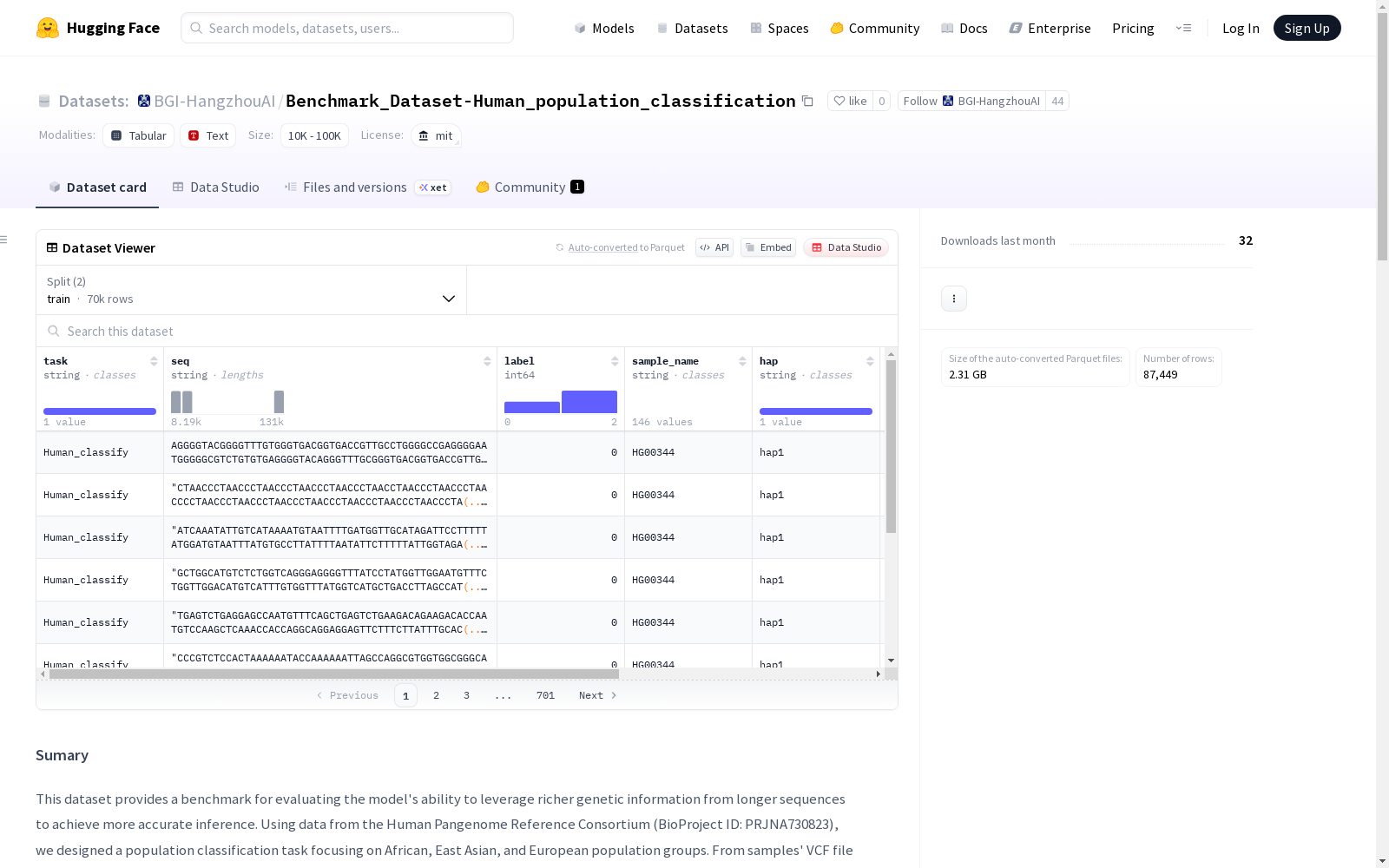
该数据集是一个用于评估模型在利用更长序列中的遗传信息进行人口群体分类的能力的基准数据集。它基于人类泛基因组参考联盟的数据,专注于非洲、东亚和欧洲三个人口群体。数据集包含了从第9号染色体的变异密集区域提取的8K、32K和128K三种长度的样本伪序列。数据预处理包括伪序列生成、变异区域统计、着丝粒移除和数据选择,并以JSONL格式保存,每个文件包含DNA序列字符串和三类人口群体的分类标签。
创建时间:
2025-10-16
原始信息汇总
数据集概述
基本信息
- 数据集名称: Benchmark_Dataset-Human_population_classification
- 许可证: MIT
- 核心用途: 评估模型利用更长序列中更丰富的遗传信息实现更准确推断的能力
数据来源与任务设计
- 数据来源于人类泛基因组参考联盟(BioProject ID: PRJNA730823)
- 专注于非洲、东亚和欧洲人群分类任务
- 从样本VCF文件和参考基因组序列生成样本伪序列
- 基于VCF文件中记录的变异位点信息,从9号染色体提取变异密集区域
序列规格
- 8,192 bp (8K)
- 32,768 bp (32K)
- 131,072 bp (128K)
基准任务详情
任务配置
| 任务名称 | 输入字段 | 训练序列数 | 验证序列数 | 测试序列数 |
|---|---|---|---|---|
| Human_population_classification_8192 | {seq, label} | 23,172 | 2,906 | 2,916 |
| Human_population_classification_32768 | {seq, label} | 23,207 | 2,913 | 2,925 |
| Human_population_classification_131072 | {seq, label} | 23,623 | 2,830 | 2,957 |
人群分类标签
| 人群 | 样本数量 | 标签 |
|---|---|---|
| EUR-European | 30 | 0 |
| AFR-African | 69 | 1 |
| EAS-East Asian | 50 | 2 |
数据处理流程
伪序列生成
- 使用
bcftools从样本VCF文件和参考基因组序列生成伪序列(包括hap1和hap2)
变异区域统计
- 使用样本VCF文件,在参考基因组9号染色体起始位置应用三种不同长度的滑动窗口(8K、32K、128K)
- 连续窗口之间的重叠为窗口长度的一半
- 统计每个窗口内的变异数量,按变异计数降序排列窗口以识别变异密集的基因组坐标
着丝粒去除
- 根据BED文件过滤掉重复和非编码的着丝粒区域
- 生成基因组窗口到对应变异计数的最终映射
数据选择
- 每个标签的样本按8:1:1的比例分为训练集、验证集和测试集
- 基于先前获得的窗口-变异计数映射,对每个样本的9号染色体hap1伪序列进行分割
- 从最高变异计数开始向下选择区域,同时确保每个标签的序列数量大致平衡
数据格式
- 数据集以JSONL格式保存
- 每条记录包含:
"seq": DNA序列字符串(A/C/G/T,大写)"label": 三元分类指示器(0 = CEU,1 = AFR,2 = EAS)
模型信息
- 使用XGBoost分类器进行个体序列的分类预测
- XGBoost模型仅使用训练集和测试集
- 保留的验证集可供需要超参数调优的算法(如多层感知机MLP)使用
搜集汇总
数据集介绍
构建方式
在人类群体遗传学研究中,准确解析基因组变异对理解种群演化具有重要意义。该数据集基于人类泛基因组参考联盟的原始测序数据,通过生物信息学流程构建而成:首先利用bcftools工具将样本VCF文件与参考基因组序列整合生成伪序列;随后针对9号染色体变异密集区域,采用三种不同长度的滑动窗口(8K/32K/128K)进行序列截取,并通过变异数量排序筛选出信息量最丰富的基因组区间;最后通过严格的质量控制流程,剔除着丝粒等重复区域,确保序列数据的生物学有效性。
特点
该数据集的设计充分考虑了基因组序列分析的技术需求,其显著特征体现在三个方面:涵盖非洲、东亚和欧洲三大地理人群的典型代表,通过0/1/2标签实现清晰的三元分类体系;提供三种不同长度的序列规格,为研究序列长度与分类性能的关联性提供实验基础;采用8:1:1的标准比例划分训练集、验证集和测试集,每个长度规格均包含两万余条训练序列,保障模型训练的统计效力。
使用方法
为便于研究人员开展对比实验,数据集采用模块化组织架构。通过HuggingFace数据集的标准接口,用户既可完整下载全部数据,也可按需加载特定长度的分类任务。在具体应用中,开发者可通过指定task_name参数调用8K/32K/128K任一子集,每个样本以JSONL格式提供DNA序列字符串和分类标签,这种设计既支持XGBoost等传统机器学习算法,也为需要超参数调优的神经网络模型预留了验证集接口。
背景与挑战
背景概述
人类基因组学研究进入后基因组时代,基于群体遗传变异的精细分类成为精准医学的重要基础。该数据集由华大基因人工智能团队于2023年构建,依托人类泛基因组参考联盟的原始数据,聚焦非洲、东亚和欧洲三大群体分类任务。通过提取9号染色体高变异密度区域,构建了8K至128K三种长度的DNA伪序列,采用XGBoost分类器实现群体溯源,为探索长序列遗传信息在群体遗传学中的应用提供了标准化评估框架。
当前挑战
在群体遗传分类领域,如何从海量单核苷酸多态性中提取具有群体区分度的特征仍是核心难题。数据集构建过程中面临多重挑战:需通过滑动窗口策略平衡序列长度与变异密度,规避着丝粒等重复区域对分类的干扰;在保持三类群体样本量均衡的同时,还需确保训练集与测试集在遗传结构上的独立性,这对后续模型的泛化能力提出更高要求。
常用场景
经典使用场景
在群体遗传学研究中,该数据集通过构建非洲、东亚和欧洲人群的遗传变异分类任务,为评估模型处理长序列遗传信息的能力提供了标准化基准。其核心应用场景聚焦于利用不同长度(8K至128K碱基对)的染色体9变异密集区域伪序列,通过XGBoost分类器实现跨人群遗传特征的精准判别,为基因组序列分析提供了可复现的实验框架。
实际应用
在医学基因组学领域,该数据集支撑的种群分类模型可直接应用于精准医疗中的药物基因组学分析,通过个体遗传背景预测提升用药安全性。同时为法医遗传学中的生物地理祖先推断提供技术验证平台,其多尺度序列设计更适用于临床基因检测仪器的算法优化与性能校准。
衍生相关工作
基于该数据集的基准特性,已衍生出多项深度学习方法在群体遗传学中的创新应用。例如采用多层感知机进行超参数优化的分类模型,以及结合图神经网络的跨人群变异关联分析。这些工作进一步推动了端到端基因组序列分析框架的发展,为构建新一代人群遗传结构图谱奠定了算法基础。
以上内容由遇见数据集搜集并总结生成


