MAP格式
收藏arXiv2025-05-06 更新2025-05-08 收录
下载链接:
http://webs.iiitd.edu.in/raghava/maprepo/
下载链接
链接失效反馈官方服务:
资源简介:
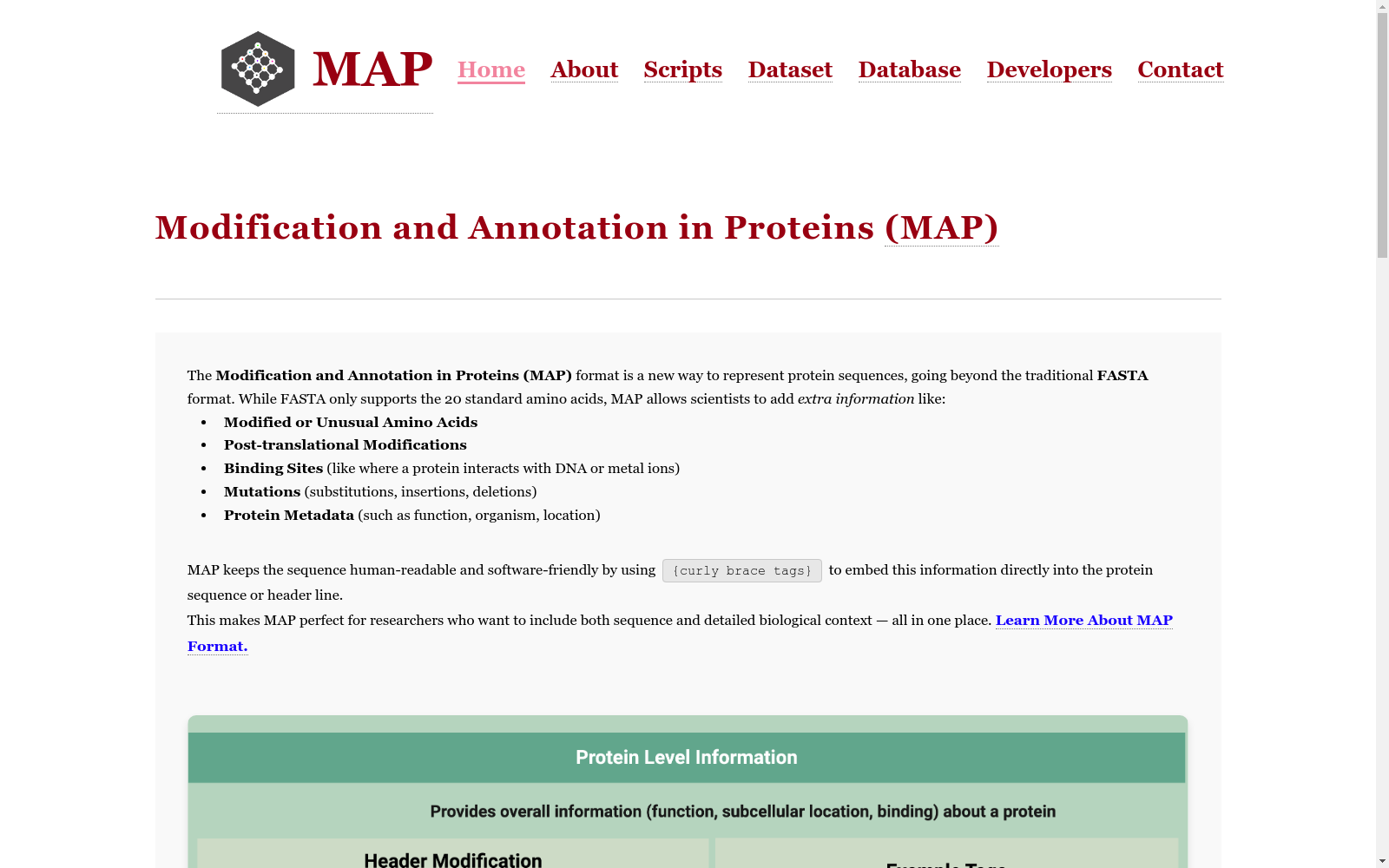
MAP格式是一个新的蛋白质序列表示方法,它扩展了FASTA格式,使其能够包含化学修饰和非自然残基,以及结构注释和突变信息。该格式在序列中嵌入修改标记和注释,同时在标题行中使用结构化标签来表示蛋白质级别的元数据。MAP格式的设计目标是捕获残基级别的修饰,包括化学修饰、非标准氨基酸、结合位点和突变,并在标题中包含蛋白质级别的描述符,例如来源生物体、功能类别和数据库ID。该格式旨在保持易读性,并与现有工具兼容。
The MAP format is a novel protein sequence representation method that extends the FASTA format to enable the inclusion of chemical modifications, non-natural residues, structural annotations, and mutation information. This format embeds modification markers and annotations within the sequence, while using structured tags in the header line to represent protein-level metadata. The design goal of the MAP format is to capture residue-level modifications, including chemical modifications, non-standard amino acids, binding sites, and mutations, and include protein-level descriptors such as source organisms, functional categories, and database IDs in the header. This format is designed to maintain readability and compatibility with existing tools.
提供机构:
印德拉普拉斯特哈信息技术学院计算生物学系
创建时间:
2025-05-06
搜集汇总
数据集介绍
构建方式
MAP格式是一种扩展的FASTA格式,旨在全面注释蛋白质序列中的化学修饰、突变和结构特征。其构建方式通过在FASTA头行引入元标签进行蛋白质级别的注释,并在序列行内使用内联标签标记残基级别的修饰。标准单字母氨基酸代码被扩展为包含花括号标签,用于表示磷酸化、乙酰化、非天然残基、环化等多种修饰。头行元数据还包含生物体、功能和序列变体等信息。该格式的设计目标包括保留FASTA的简洁性和人类可读性,同时增强注释能力。
特点
MAP格式的主要特点包括其灵活性和兼容性。它不仅支持传统的氨基酸序列表示,还能通过内联标签直接嵌入残基级别的修饰信息,如磷酸化、糖基化等。头行元标签提供标准化的蛋白质描述,便于程序化解析。MAP格式保留了FASTA的核心结构,确保与现有生物信息学工具的兼容性。此外,其设计允许通过简单去除标签转换为标准FASTA序列,便于在传统流程中使用。这种格式特别适用于蛋白质治疗领域,能够精确描述工程化蛋白质的复杂修饰。
使用方法
MAP格式的使用方法包括直接编辑和程序化处理。研究人员可以使用文本编辑器手动添加或修改注释标签,标签紧跟在相关残基之后。对于大规模数据分析,开发了专门的解析器来读取和写入MAP条目,并能剥离所有注释标签生成标准FASTA序列。该格式适用于多种应用场景,如蛋白质数据库管理、结构生物学分析和治疗性蛋白质设计。MAP格式文件可通过提供的Python脚本或在线工具与其他格式(如PDB)相互转换,便于集成到现有工作流程中。
背景与挑战
背景概述
MAP格式是由印度信息技术研究所德里分校的计算生物学系Gajendra P. S. Raghava教授团队于近年提出的蛋白质序列表示新标准。该格式旨在解决传统FASTA格式在表示化学修饰、非天然氨基酸残基及蛋白质变体时的局限性。作为FASTA格式的扩展,MAP通过引入元标签和内联注释机制,实现了对磷酸化、乙酰化、环化等多种修饰类型的精确标注,同时保持了良好的人类可读性。这一创新为蛋白质组学、药物研发等领域提供了更丰富的数据表示框架,特别是在蛋白质治疗剂等前沿研究方向展现出重要应用价值。
当前挑战
MAP格式面临的核心挑战主要体现在三个方面:在领域问题层面,需要解决复杂蛋白质变体(如多链二硫键连接蛋白)的线性表示难题,现有方法难以直观描述非连续序列间的结构关系;在构建过程中,缺乏统一的受控词汇表导致修饰标注存在不一致性风险,如磷酸化可能被标注为{ptm:Phos}或{ptm:Phospho};在技术实现上,注释标签的引入增加了序列字符串长度,可能影响序列比对、 motif搜索等分析结果的准确性。此外,该格式需要与主流生物信息学工具(如BioPython)集成以实现广泛采用,这也是当前亟待突破的工程挑战。
常用场景
经典使用场景
MAP格式在蛋白质序列注释和化学修饰表示领域具有广泛的应用,尤其在蛋白质组学和生物信息学研究中表现突出。该格式通过扩展FASTA格式,支持在蛋白质序列中直接嵌入化学修饰、非天然氨基酸残基以及结构注释等信息。其经典使用场景包括蛋白质数据库的构建和维护,如UniProt和PDB数据库,研究者可以利用MAP格式高效地存储和交换包含复杂修饰的蛋白质序列数据。
解决学术问题
MAP格式解决了传统FASTA格式无法表示化学修饰和非天然氨基酸残基的局限性,为蛋白质序列的全面注释提供了标准化工具。通过引入内联标签和元数据标签,MAP格式能够精确描述磷酸化、乙酰化、环化等修饰,以及突变和序列变体。这一创新显著提升了蛋白质序列数据的表达能力和信息密度,为蛋白质功能预测、结构分析和药物设计等研究提供了可靠的数据基础。
衍生相关工作
围绕MAP格式,研究者开发了一系列衍生工具和资源,如MAPrepo数据库和配套的Python脚本库。这些资源进一步扩展了MAP格式的应用范围,包括蛋白质结构预测工具(如PepStrMOD)和修饰位点预测算法(如GlycoEP)。此外,MAP格式的标准化设计也启发了其他序列表示方法的改进,如PEFF格式的进一步发展,推动了蛋白质注释领域的整体进步。
以上内容由遇见数据集搜集并总结生成


