Tahoe-100M
收藏魔搭社区2025-12-05 更新2025-11-15 收录
下载链接:
https://modelscope.cn/datasets/tahoebio/Tahoe-100M
下载链接
链接失效反馈官方服务:
资源简介:
# Tahoe-100M
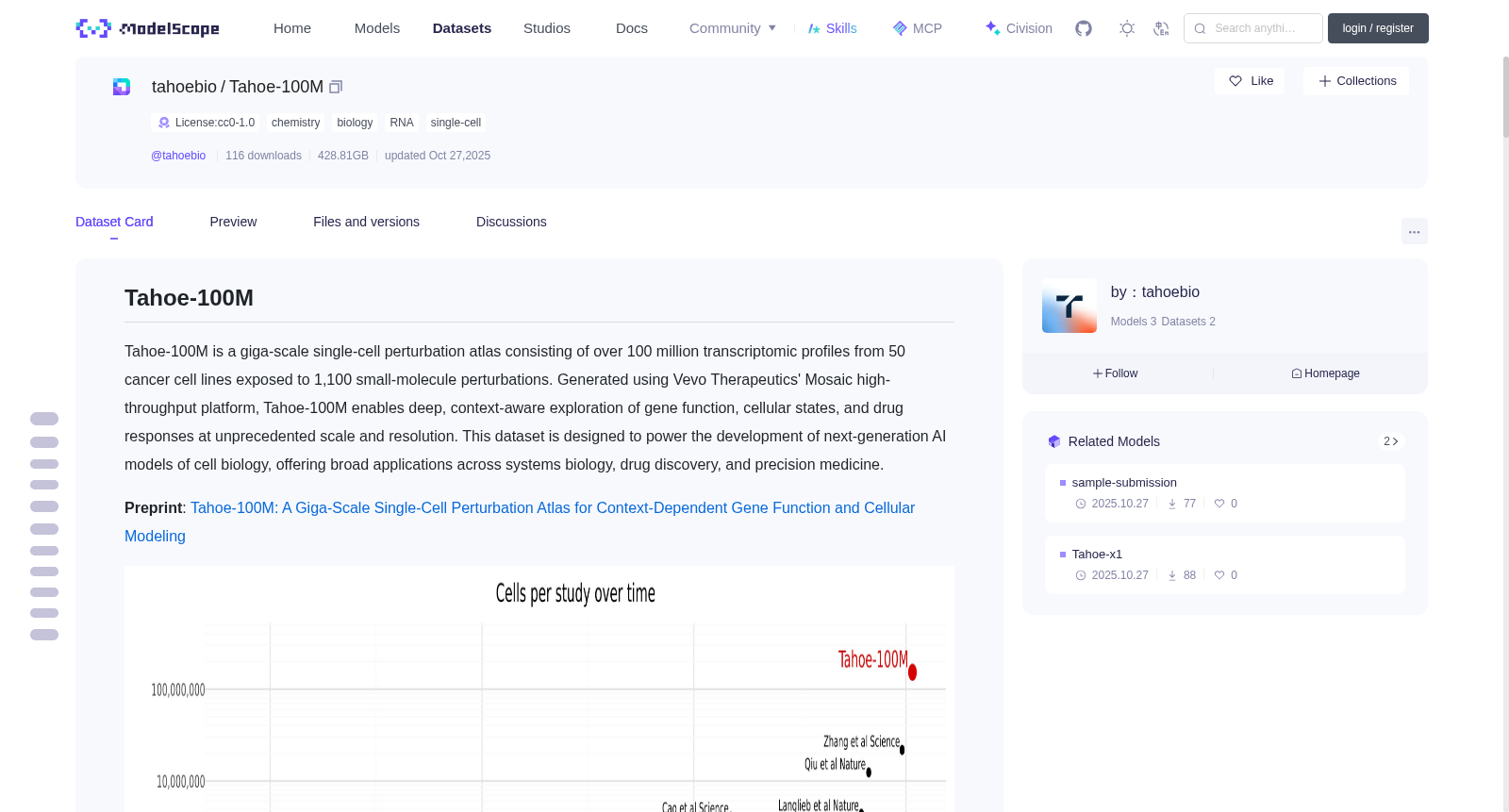
Tahoe-100M is a giga-scale single-cell perturbation atlas consisting of over 100 million transcriptomic profiles from
50 cancer cell lines exposed to 1,100 small-molecule perturbations. Generated using Vevo Therapeutics'
Mosaic high-throughput platform, Tahoe-100M enables deep, context-aware exploration of gene function, cellular states, and drug responses at unprecedented scale and resolution.
This dataset is designed to power the development of next-generation AI models of cell biology,
offering broad applications across systems biology, drug discovery, and precision medicine.
**Preprint**: [Tahoe-100M: A Giga-Scale Single-Cell Perturbation Atlas for Context-Dependent Gene Function and Cellular Modeling](https://www.biorxiv.org/content/10.1101/2025.02.20.639398v1)
<img src="https://pbs.twimg.com/media/Gkpp8RObkAM-fxe?format=jpg&name=4096x4096" width="1024" height="1024">
## Quickstart
```python
from datasets import load_dataset
# Load dataset in streaming mode
ds = load_dataset("tahoebio/Tahoe-100m", streaming=True, split="train")
# View the first record
next(ds.iter(1))
```
### Tutorials
Please refer to our tutorials for examples on using the data, accessing metadata tables and converting to/from the anndata format.
Please see the [Data Loading Tutorial](tutorials/loading_data.ipynb) for a walkthrough on using the data.
<table>
<thead>
<tr>
<th>Notebook</th>
<th>URL</th>
<th>Colab</th>
</tr>
</thead>
<tbody>
<tr>
<td>Loading the dataset from huggingface, accessing metadata, mapping to anndata</td>
<td>
<a href="https://huggingface.co/datasets/tahoebio/Tahoe-100M/blob/main/tutorials/loading_data.ipynb" target="_blank">
Link
</a>
</td>
<td>
<a href="https://colab.research.google.com/#fileId=https://huggingface.co/datasets/tahoebio/Tahoe-100M/blob/main/tutorials/loading_data.ipynb" target="_blank">
<img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open in Colab"/>
</a>
</td>
</tr>
</tbody>
</table>
### Community Resources
Here are a links to few resources created by the community. We would love to feature additional tutorials from the community, if you have built something on top of
Tahoe-100M, please let us know and we would love to feature your work.
<table>
<thead>
<tr>
<th>Resource</th>
<th>Contributor</th>
<th>URL</th>
</tr>
</thead>
<tbody>
<tr>
<td>Analysis guide for Tahoe-100M using rapids-single-cell, scanpy and dask</td>
<td><a href="https://github.com/scverse" target="_blank">SCVERSE</a></td>
<td><a href="https://github.com/theislab/vevo_Tahoe_100m_analysis/tree/tahoe-DGX-fix" target="_blank">Link</a></td>
</tr>
<tr>
<td>Tutorial for accessing Tahoe-100M h5ad files hosted by the Arc Institute</td>
<td><a href="https://github.com/ArcInstitute" target="_blank">Arc Institute</a></td>
<td><a href="https://github.com/ArcInstitute/arc-virtual-cell-atlas/blob/main/tahoe-100M/tutorial-py.ipynb" target="_blank">Link</a></td>
</tr>
</tbody>
</table>
## Dataset Features
We provide multiple tables with the dataset including the main data (raw counts) in the `expression_data` table as well as
various metadata in the `gene_metadata`,`sample_metadata`,`drug_metadata`,`cell_line_metadata`,`obs_metadata` tables.
The main data can be downloaded as follows:
```python
from datasets import load_dataset
tahoe_100m_ds = load_dataset("tahoebio/Tahoe-100M", streaming=True, split="train")
```
Setting `stream=True` instantiates an `IterableDataset` and prevents needing to
download the full dataset first. See [tutorial](tutorials/loading_data.ipynb) for an end-to-end example.
The expression_data table has the following fields:
| **Field Name** | **Type** | **Description** |
|------------------------|-------------------|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `genes` | `sequence<int64>` | Gene identifiers (integer token IDs) corresponding to each gene with non-zero expression in the cell. This sequence aligns with the `expressions` field. The gene_metadata table can be used to map the token_IDs to gene_symbols or ensembl_IDs. The first entry for each row is just a marker token and should be ignored (See [data-loading tutorial](tutorials/loading_data.ipynb)) |
| `expressions` | `sequence<float32>` | Raw count values for each gene, aligned with the `genes` field. The first entry just marks a CLS token and should be ignored when parsing. |
| `drug` | `string` | Name of the treatment. DMSO_TF marks vehicle controls, use DMSO_TF along with plate to get plate matched controls. |
| `sample` | `string` | Unique identifier for the sample from which the cell was derived. Can be used to merge information from the `sample_metadata` table. Distinguishes replicate treatments. |
| `BARCODE_SUB_LIB_ID`| `string` | Combination of barcode and sublibary identifiers. Unique for each cell in the dataset. Can be used as an index key when referencing to the `obs_metadata` table. |
| `cell_line_id` | `string` | Unique identifier for the cancer cell line from which the cell originated. We use Cellosaurus IDs were, but additional identifiers such as DepMap IDs are provided in the `cell_line_metadata` table. |
| `moa-fine` | `string` | Fine-grained mechanism of action (MOA) annotation for the drug, specifying the biological process or molecular target affected. Derived from MedChemExpress and curated with GPT-based annotations. |
| `canonical_smiles` | `string` | Canonical SMILES (Simplified Molecular Input Line Entry System) string representing the molecular structure of the perturbing compound. |
| `pubchem_cid` | `string` | PubChem Compound Identifier for the drug, allowing cross-referencing with public chemical databases. An empty string is used for DMSO controls. Please cast to int before querrying pubchem. |
| `plate` | `string` | Identifier for the 96-well plate (1–14) in which the mixed-cell spheroid was seeded and treated. |
## Additional metadata
### Gene Metadata
```python
gene_metadata = load_dataset("taheobio/Tahoe-100M","gene_metadata", split="train")
```
| Column Name | Description |
|---------------|-------------------------------------------------------------------------------------------------------------|
| `gene_symbol` | The HGNC-approved gene symbol corresponding to each gene (e.g., *TP53*, *BRCA1*). |
| `ensembl_id` | The Ensembl gene identifier (e.g., *ENSG00000000003*) based on Ensembl release 109 and genome build 38. |
| `token_id` | An integer token ID used to represent each gene. This is the ID used in the `genes` field in the main data. |
### Sample Metadata
```python
sample_metadata = load_dataset("tahoebio/Tahoe-100M","sample_metadata", split="train")
```
The sample_metadata has additional information for aggregate quality metrics for the sample as well as the concentration.
| Column Name | Description |
|------------------------|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `sample` | Unique identifier for the sample from which the cell was derived. Unique key for this table. |
| `plate` | Identifier (1–14) for the 96-well plate for the sample |
| `mean_gene_count` | Average number of unique genes detected per cell for the given sample. |
| `mean_tscp_count` | Average number of transcripts (UMIs) detected per cell in the sample. |
| `mean_mread_count` | Average number of reads per cell. |
| `mean_pcnt_mito` | Mean percentage of total reads that map to mitochondrial genes, across cells in the sample. |
| `drug` | Name of the treatment used to perturb the cells in the sample. |
| `drugname_drugconc` | String combining the compound name, concentration and concentration unit (e.g., `[('8-Hydroxyquinoline',0.05,'uM')]`), used to uniquely label each treatment condition. |
### Drug Metadata
```python
drug_metadata = load_dataset("tahoebio/Tahoe-100M","drug_metadata", split="train")
```
The drug_metadata has additional information about each treatment.
| Column Name | Description |
|------------------------|---------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `drug` | Name of the treatment used to perturb the cells in the sample. Unique key for this table |
| `targets` | List of gene symbols representing the known molecular targets of the compound. Targets were proposed by GPT-4o based on compound names and then validated against MedChemExpress information. |
| `moa-broad` | Broad classification of the compound’s mechanism of action (MOA), typically categorized as "inhibitor/antagonist," "activator/agonist," or "unclear." GPT-4o inferred this using compound target data and curated descriptions from MedChemExpress. |
| `moa-fine` | Specific functional annotation of the compound's MOA (e.g., "Proteasome inhibitor" or "MEK inhibitor"). These fine-grained labels were selected from a curated list of 25 MOA categories and assigned by GPT-4o with validation against compound descriptions. |
| `human-approved` | Indicates whether the compound is approved for human use ("yes" or "no"). GPT-4o provided these labels using prior knowledge and validation from public sources such as clinicaltrials.gov. |
| `clinical-trials` | Indicates whether the compound has been evaluated in any registered clinical trials ("yes" or "no"). Determined using GPT-4o and corroborated using clinicaltrials.gov searches. |
| `gpt-notes-approval` | Contextual notes generated by GPT-4o summarizing the compound’s approval status, common clinical usage, or nuances such as formulation-specific approvals. |
| `canonical_smiles` | The compound's SMILES (Simplified Molecular Input Line Entry System) representation, capturing its molecular structure as a text string. |
| `pubchem_cid` | The PubChem Compound Identifier (CID), a unique numerical ID linking the compound to its entry in the PubChem database. |
### Cell Line Metadata
```python
cell_line_metadata = load_dataset("tahoebio/Tahoe-100M","cell_line_metadata", split="train")
```
The cell-line metadata table has additional information about the key driver mutations for each cell line.
| Column Name | Description |
|----------------------------------|------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `cell_name` | Standard name of the cancer cell line (e.g., *A549*). |
| `Cell_ID_DepMap` | Unique identifier for the cell line in the DepMap project (e.g., *ACH-000681*) |
| `Cell_ID_Cellosaur` | Cellosaurus accession ID (e.g., *CVCL_0023*). This is the ID used in the main dataset. |
| `Organ` | Tissue or organ of origin for the cell line (e.g., *Lung*), used to interpret lineage-specific responses and biological context. |
| `Driver_Gene_Symbol` | HGNC-approved symbol of a known or putative driver gene with functional alterations in this cell line (e.g., *KRAS*, *CDKN2A*). We report a curated list of driver mutations per cell-line. |
| `Driver_VarZyg` | Zygosity of the driver variant (e.g., *Hom* for homozygous, *Het* for heterozygous) |
| `Driver_VarType` | Type of genetic alteration (e.g., *Missense*, *Frameshift*, *Stopgain*, *Deletion*) |
| `Driver_ProtEffect_or_CdnaEffect`| Specific protein or cDNA-level annotation of the mutation (e.g., *p.G12S*, *p.Q37*), providing precise information on the variant’s consequence. |
| `Driver_Mech_InferDM` | Inferred functional mechanism of the mutation (e.g., *LoF* for loss-of-function, *GoF* for gain-of-function) |
| `Driver_GeneType_DM` | Classification of the driver gene as an *Oncogene* or *Suppressor* |
## Citation
Please cite:
```
@article{zhang2025tahoe,
title={Tahoe-100M: A Giga-Scale Single-Cell Perturbation Atlas for Context-Dependent Gene Function and Cellular Modeling},
author={Zhang, Jesse and Ubas, Airol A and de Borja, Richard and Svensson, Valentine and Thomas, Nicole and Thakar, Neha and Lai, Ian and Winters, Aidan and Khan, Umair and Jones, Matthew G and others},
journal={bioRxiv},
pages={2025--02},
year={2025},
publisher={Cold Spring Harbor Laboratory}
}
```
# Tahoe-100M
Tahoe-100M 是一个十亿级规模的单细胞扰动图谱(single-cell perturbation atlas),包含来自50种癌细胞系、经1100种小分子扰动处理后的超过1亿条转录组谱。该数据集依托Vevo Therapeutics的Mosaic高通量平台构建,能够以前所未有的规模与分辨率,实现对基因功能、细胞状态及药物反应的深度情境感知探索。本数据集旨在推动下一代细胞生物学人工智能模型的研发,可广泛应用于系统生物学、药物发现与精准医学等领域。
**预印本**:[Tahoe-100M: 面向情境依赖基因功能与细胞建模的十亿级单细胞扰动图谱](https://www.biorxiv.org/content/10.1101/2025.02.20.639398v1)
<img src="https://pbs.twimg.com/media/Gkpp8RObkAM-fxe?format=jpg&name=4096x4096" width="1024" height="1024">
## 快速入门
python
from datasets import load_dataset
# 以流式加载模式加载数据集
ds = load_dataset("tahoebio/Tahoe-100m", streaming=True, split="train")
# 查看第一条数据记录
next(ds.iter(1))
### 教程
请参阅我们的教程以了解数据使用、元数据表访问以及与anndata格式互转的示例。如需了解数据使用的完整流程,请参阅[数据加载教程](tutorials/loading_data.ipynb)。
<table>
<thead>
<tr>
<th>教程文档</th>
<th>链接地址</th>
<th>Colab运行地址</th>
</tr>
</thead>
<tbody>
<tr>
<td>从Hugging Face加载数据集、访问元数据并映射至anndata格式</td>
<td>
<a href="https://huggingface.co/datasets/tahoebio/Tahoe-100M/blob/main/tutorials/loading_data.ipynb" target="_blank">
链接
</a>
</td>
<td>
<a href="https://colab.research.google.com/#fileId=https://huggingface.co/datasets/tahoebio/Tahoe-100M/blob/main/tutorials/loading_data.ipynb" target="_blank">
<img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open in Colab"/>
</a>
</td>
</tr>
</tbody>
</table>
### 社区资源
以下为社区贡献的部分资源。我们欢迎展示更多基于Tahoe-100M开发的社区教程,若您有相关成果,欢迎联系我们进行展示。
<table>
<thead>
<tr>
<th>资源内容</th>
<th>贡献方</th>
<th>链接地址</th>
</tr>
</thead>
<tbody>
<tr>
<td>使用rapids-single-cell、scanpy与dask进行Tahoe-100M分析的指南</td>
<td><a href="https://github.com/scverse" target="_blank">SCVERSE</a></td>
<td><a href="https://github.com/theislab/vevo_Tahoe_100m_analysis/tree/tahoe-DGX-fix" target="_blank">链接</a></td>
</tr>
<tr>
<td>访问Arc Institute托管的Tahoe-100M h5ad文件的教程</td>
<td><a href="https://github.com/ArcInstitute" target="_blank">Arc Institute</a></td>
<td><a href="https://github.com/ArcInstitute/arc-virtual-cell-atlas/blob/main/tahoe-100M/tutorial-py.ipynb" target="_blank">链接</a></td>
</tr>
</tbody>
</table>
## 数据集特征
本数据集提供多张数据表,包括存储原始计数的主数据表`expression_data`,以及`gene_metadata`、`sample_metadata`、`drug_metadata`、`cell_line_metadata`、`obs_metadata`等元数据表。
主数据可通过以下方式下载:
python
from datasets import load_dataset
tahoe_100m_ds = load_dataset("tahoebio/Tahoe-100M", streaming=True, split="train")
设置`stream=True`将实例化一个`IterableDataset`(可迭代数据集),无需预先下载完整数据集。如需完整示例,请参阅[教程](tutorials/loading_data.ipynb)。
`expression_data`表包含以下字段:
| **字段名** | **数据类型** | **说明** |
|------------------------|-------------------|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `genes` | `sequence<int64>` | 基因标识符(整数Token ID),对应细胞中表达量非零的基因。该序列与`expressions`字段对齐。可通过`gene_metadata`表将Token ID映射为基因符号或Ensembl ID。每行的首个条目仅为标记Token,需忽略(详见[数据加载教程](tutorials/loading_data.ipynb)) |
| `expressions` | `sequence<float32>` | 各基因的原始计数,与`genes`字段对齐。解析时需忽略首个条目,该条目仅用于标记CLS Token。 |
| `drug` | `string` | 处理剂名称。`DMSO_TF`代表溶剂对照,可结合`plate`字段获取板匹配的对照组。 |
| `sample` | `string` | 细胞来源样本的唯一标识符,可用于关联`sample_metadata`表中的信息,区分重复处理组。 |
| `BARCODE_SUB_LIB_ID`| `string` | 条形码与子文库标识符的组合,为数据集中每个细胞的唯一标识,可作为索引键关联`obs_metadata`表。 |
| `cell_line_id` | `string` | 细胞来源癌细胞系的唯一标识符。本数据集使用Cellosaurus ID,同时`cell_line_metadata`表中提供了DepMap ID等额外标识符。 |
| `moa-fine` | `string` | 药物的精细作用机制(MOA)注释,明确标注其影响的生物学过程或分子靶点。数据源自MedChemExpress,并经基于GPT的注释进行人工校验。 |
| `canonical_smiles` | `string` | 表示扰动化合物分子结构的标准化简化分子线性输入系统(Simplified Molecular Input Line Entry System,简称SMILES)字符串。 |
| `pubchem_cid` | `string` | 药物的PubChem化合物标识符,可用于跨公共化学数据库交叉引用。DMSO对照组使用空字符串。查询PubChem前请先将其转换为整数类型。 |
| `plate` | `string` | 96孔板(编号1–14)的标识符,混合细胞球体在此板中接种并接受处理。 |
## 额外元数据
### 基因元数据
python
gene_metadata = load_dataset("taheobio/Tahoe-100M","gene_metadata", split="train")
| 列名 | 说明 |
|---------------|-------------------------------------------------------------------------------------------------------------|
| `gene_symbol` | 对应基因的HGNC官方批准基因符号(例如*TP53*、*BRCA1*)。 |
| `ensembl_id` | 基于Ensembl版本109和基因组版本38的Ensembl基因标识符(例如*ENSG00000000003*)。 |
| `token_id` | 用于表示每个基因的整数Token ID,即主数据中`genes`字段使用的ID。 |
### 样本元数据
python
sample_metadata = load_dataset("tahoebio/Tahoe-100M","sample_metadata", split="train")
`sample_metadata`表包含样本的聚合质量指标与浓度信息。
| 列名 | 说明 |
|------------------------|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `sample` | 细胞来源样本的唯一标识符,为本表的唯一键。 |
| `plate` | 对应样本的96孔板标识符(1–14) |
| `mean_gene_count` | 给定样本中每个细胞检测到的独特基因的平均数量。 |
| `mean_tscp_count` | 样本中每个细胞检测到的唯一分子标识符(Unique Molecular Identifiers,简称UMIs)的平均数量。 |
| `mean_mread_count` | 每个细胞的平均读取数。 |
| `mean_pcnt_mito` | 样本中所有细胞的线粒体基因映射读取数占总读取数的平均百分比。 |
| `drug` | 用于扰动样本中细胞的处理剂名称。 |
| `drugname_drugconc` | 结合化合物名称、浓度与浓度单位的字符串(例如`[('8-Hydroxyquinoline',0.05,'uM')]`),用于唯一标记每个处理条件。 |
### 药物元数据
python
drug_metadata = load_dataset("tahoebio/Tahoe-100M","drug_metadata", split="train")
`drug_metadata`表包含每种处理剂的额外信息。
| 列名 | 说明 |
|------------------------|---------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `drug` | 用于扰动样本中细胞的处理剂名称,为本表的唯一键 |
| `targets` | 代表化合物已知分子靶点的基因符号列表。靶点由GPT-4o基于化合物名称提出,后经MedChemExpress信息验证。 |
| `moa-broad` | 化合物作用机制(MOA)的宽泛分类,通常分为“抑制剂/拮抗剂”“激活剂/激动剂”或“未明确”。由GPT-4o基于化合物靶点数据与MedChemExpress的人工 curated 描述推断。 |
| `moa-fine` | 化合物MOA的具体功能注释(例如“蛋白酶体抑制剂”或“MEK抑制剂”)。这些精细标签选自25个人工 curated MOA类别,由GPT-4o分配并经化合物描述验证。 |
| `human-approved` | 指示化合物是否获批用于人类使用(“yes”或“no”)。由GPT-4o基于先验知识并经clinicaltrials.gov等公共来源验证。 |
| `clinical-trials` | 指示化合物是否已在任何注册临床试验中进行评估(“yes”或“no”)。由GPT-4o确定,并经clinicaltrials.gov搜索佐证。 |
| `gpt-notes-approval` | GPT-4o生成的上下文注释,总结化合物的获批状态、常见临床用途或剂型特异性获批等细节。 |
| `canonical_smiles` | 化合物的SMILES表示,以文本字符串形式捕获其分子结构。 |
| `pubchem_cid` | PubChem化合物标识符(CID),是将化合物链接到PubChem数据库条目的唯一数值ID。 |
### 细胞系元数据
python
cell_line_metadata = load_dataset("tahoebio/Tahoe-100M","cell_line_metadata", split="train")
`cell-line`元数据表包含每种细胞系的关键驱动突变信息。
| 列名 | 说明 |
|----------------------------------|------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| `cell_name` | 癌细胞系的标准名称(例如*A549*)。 |
| `Cell_ID_DepMap` | DepMap项目中细胞系的唯一标识符(例如*ACH-000681*) |
| `Cell_ID_Cellosaur` | Cellosaurus登录ID(例如*CVCL_0023*),即主数据集中使用的ID。 |
| `Organ` | 细胞系的组织或器官来源(例如*肺*),用于解析谱系特异性反应与生物学背景。 |
| `Driver_Gene_Symbol` | 该细胞系中存在功能改变的已知或推定驱动基因的HGNC批准符号(例如*KRAS*、*CDKN2A*)。我们提供了每个细胞系的人工 curated 驱动突变列表。 |
| `Driver_VarZyg` | 驱动变异的合子性(例如*Hom*代表纯合子,*Het*代表杂合子) |
| `Driver_VarType` | 遗传改变的类型(例如*错义突变*、*移码突变*、*无义突变*、*缺失*) |
| `Driver_ProtEffect_or_CdnaEffect`| 突变的具体蛋白质或cDNA水平注释(例如*p.G12S*、*p.Q37*),提供变异后果的精确信息。 |
| `Driver_Mech_InferDM` | 推断的突变功能机制(例如*LoF*代表功能丧失,*GoF*代表功能获得) |
| `Driver_GeneType_DM` | 驱动基因的分类,分为*致癌基因*或*抑癌基因* |
## 引用
请引用:
@article{zhang2025tahoe,
title={Tahoe-100M: A Giga-Scale Single-Cell Perturbation Atlas for Context-Dependent Gene Function and Cellular Modeling},
author={Zhang, Jesse and Ubas, Airol A and de Borja, Richard and Svensson, Valentine and Thomas, Nicole and Thakar, Neha and Lai, Ian and Winters, Aidan and Khan, Umair and Jones, Matthew G and others},
journal={bioRxiv},
pages={2025--02},
year={2025},
publisher={Cold Spring Harbor Laboratory}
}
提供机构:
maas
创建时间:
2025-10-27
搜集汇总
数据集介绍
背景与挑战
背景概述
Tahoe-100M是一个包含超过1亿个转录组数据的大规模单细胞扰动图谱,覆盖50种癌细胞系和1,100种小分子扰动。该数据集支持系统生物学、药物发现和精准医学的研究,提供了丰富的元数据和原始计数数据。
以上内容由遇见数据集搜集并总结生成


