clinvar_multiz_100_partial_bwa_align
收藏Hugging Face2025-12-02 更新2025-12-03 收录
下载链接:
https://huggingface.co/datasets/emarro/clinvar_multiz_100_partial_bwa_align
下载链接
链接失效反馈官方服务:
资源简介:
该数据集是通过bwa-mem2比对工具,将人类ClinVar数据库中的CDS区域变异的128bp序列与Multiz-100多物种序列比对得到的。数据集包含了与比对相关的多种生物的序列信息,以及变异标签和染色体位置信息。数据集分为训练集和测试集。
创建时间:
2025-12-02
原始信息汇总
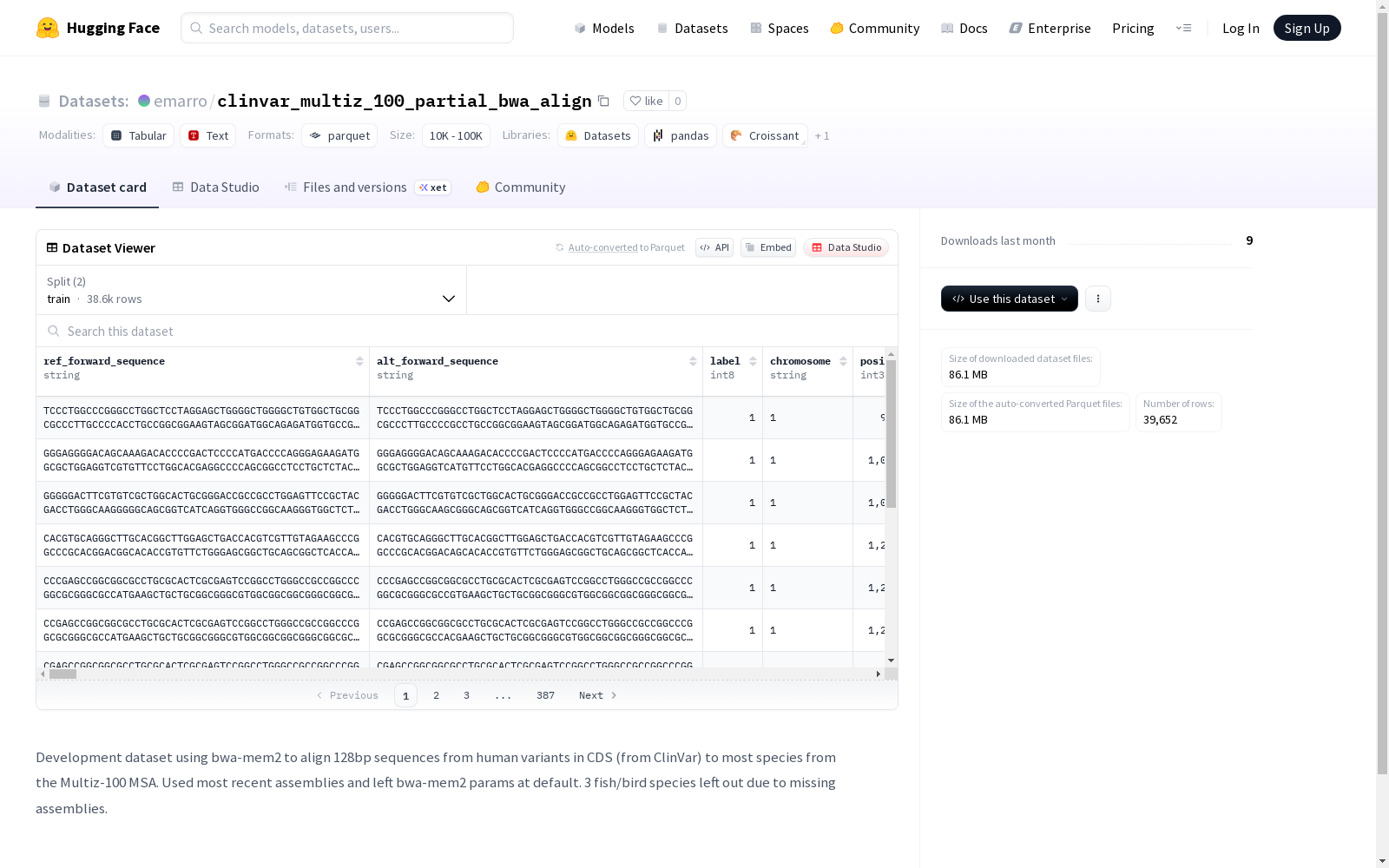
数据集概述
数据集基本信息
- 数据集名称: clinvar_multiz_100_partial_bwa_align
- 存储地址: https://huggingface.co/datasets/emarro/clinvar_multiz_100_partial_bwa_align
- 开发目的: 使用bwa-mem2将来自ClinVar的人类CDS区域变异体的128bp序列与Multiz-100多序列比对中的大多数物种进行比对。
- 技术细节: 使用了最新的基因组组装,并保持bwa-mem2参数为默认设置。由于缺少组装,遗漏了3个鱼类/鸟类物种。
数据集结构与规模
数据划分
- 训练集 (train):
- 样本数量: 38,634
- 数据大小: 495,075,113 字节
- 测试集 (test):
- 样本数量: 1,018
- 数据大小: 13,044,652 字节
整体规模
- 下载大小: 86,069,879 字节
- 数据集总大小: 508,119,765 字节
数据特征
数据集包含以下字段:
核心变异信息
ref_forward_sequence: (字符串) 参考正向序列alt_forward_sequence: (字符串) 替代正向序列label: (int8) 标签chromosome: (字符串) 染色体position: (int32) 位置
物种比对序列
数据集包含99个不同物种的比对序列字段,每个字段均为字符串类型,命名格式为[物种代号]_align。部分示例如下:
ochPri3_alignailMel1_aligntupChi1_alignoryCun2_alignfr3_align- ... (共99个物种比对字段)
配置信息
- 默认配置名称: default
- 数据文件路径:
- 训练集:
data/train-* - 测试集:
data/test-*
- 训练集:
搜集汇总
数据集介绍
构建方式
在基因组学与生物信息学领域,精准的序列比对是解析遗传变异功能的关键。该数据集以ClinVar数据库中的人类编码序列变异为起点,从中提取128bp的参考与替代序列片段。借助bwa-mem2这一高效比对工具,将这些人类变异序列与Multiz-100多物种比对所涵盖的绝大多数物种基因组进行默认参数下的全局比对。构建过程中,仅因基因组组装缺失而排除了三种鱼类与鸟类物种,最终形成了包含参考序列、替代序列、变异标签及跨物种比对信息的结构化数据。
特点
该数据集的核心特征在于其广泛的多物种覆盖与精细的序列表示。它不仅提供了人类变异位点的染色体位置与标签,更囊括了从灵长类到鱼类、哺乳类到爬行类等近百个物种的比对序列,形成了一套跨物种进化保守性分析的宝贵资源。每个样本均以固定长度的序列片段呈现,确保了机器学习模型输入的规范性。数据集划分为训练集与测试集,规模适中,兼顾了模型训练的效率与评估的可靠性,为深度学习方法在基因组学中的应用提供了标准化基准。
使用方法
该数据集主要服务于开发与评估用于遗传变异致病性预测或功能注释的计算模型。研究人员可直接加载数据集中预设的训练与测试分割,利用参考序列、替代序列以及丰富的跨物种比对序列作为模型输入特征。其提供的二分类标签适用于监督学习任务,例如训练卷积神经网络或注意力机制模型来学习序列模式与临床意义之间的关联。通过整合多物种比对信息,模型能够捕捉进化约束特征,从而提升对罕见变异或新发变异进行功能预测的泛化能力与生物学解释性。
背景与挑战
背景概述
在基因组学与生物信息学领域,精准解读人类遗传变异对疾病关联的影响,一直是推动精准医学发展的核心议题。ClinVar_multiz_100_partial_bwa_align数据集应运而生,其构建依托于ClinVar这一权威的公共变异数据库,整合了多物种比对信息,旨在通过跨物种序列保守性分析,深化对编码区变异功能效应的理解。该数据集由研究团队利用bwa-mem2工具,将人类CDS区域的128bp变异序列与Multiz-100多序列比对中的近百个物种基因组进行比对生成,体现了计算基因组学在整合进化信息以辅助临床变异解读方面的前沿探索。
当前挑战
该数据集致力于应对遗传变异致病性预测这一复杂问题,其核心挑战在于如何有效利用跨物种进化保守性特征,区分致病变异与良性多态性,尤其在面对功能效应微弱或语境依赖的变异时,模型泛化能力常受限制。在构建过程中,挑战主要体现在多物种基因组数据的整合与对齐上:不同物种的基因组组装质量参差不齐,部分物种因缺乏最新组装版本而被排除,且默认参数的bwa-mem2比对可能未优化物种特异性偏差,这可能导致比对错误或信息丢失,影响后续分析的可靠性。
常用场景
经典使用场景
在基因组学与生物信息学领域,ClinVar_multiz_100_partial_bwa_align数据集为研究人类编码序列变异的功能影响提供了关键资源。该数据集通过bwa-mem2算法将ClinVar中的人类变异序列与Multiz-100多物种比对中的近百个物种基因组进行对齐,构建了包含参考序列、变异序列及跨物种比对信息的结构化数据。其经典使用场景集中于训练和评估机器学习模型,特别是深度学习架构,以预测单核苷酸变异(SNV)或小片段插入缺失(Indel)的致病性。研究人员利用该数据集中的跨物种保守性特征,能够有效捕捉进化约束信息,从而提升变异分类的准确性,为高通量基因组注释提供可靠基础。
解决学术问题
该数据集致力于解决基因组学中一个核心学术问题:如何准确、高效地评估人类遗传变异的临床意义。传统方法依赖实验验证或保守性评分,往往耗时且覆盖有限。本数据集通过整合ClinVar的临床变异注释与跨物种多序列比对,为计算模型提供了丰富的进化背景特征,使得模型能够学习变异位点在进化过程中的保守模式。这显著推进了致病性预测的自动化研究,降低了假阳性率,并为理解变异功能机制提供了数据驱动的新途径,对精准医学和遗传疾病诊断具有深远影响。
衍生相关工作
围绕该数据集,已衍生出多项经典研究工作。例如,基于深度学习的变异致病性预测模型如EVE和PrimateAI,借鉴了其跨物种比对思路,进一步优化了特征提取架构。一些研究利用该数据集的比对信息开发了新的进化保守性评分,如phyloP和GERP的改进版本。同时,在迁移学习领域,该数据集被用作预训练数据,以提升模型在稀缺变异类型上的泛化能力。这些工作共同推动了计算基因组学的发展,为变异解读建立了更稳健的算法基础。
以上内容由遇见数据集搜集并总结生成


