FoldBench
收藏github2025-05-31 更新2025-06-01 收录
下载链接:
https://github.com/BEAM-Labs/FoldBench
下载链接
链接失效反馈官方服务:
资源简介:
FoldBench是一个低同源性的基准测试集,涵盖了蛋白质、核酸、配体等多种生物分子结构,用于生物分子结构预测的评估。
FoldBench is a low-homology benchmark dataset covering diverse biomolecular structures including proteins, nucleic acids, ligands and other related biomolecules, which is used for the evaluation of biomolecular structure prediction tasks.
创建时间:
2025-05-22
原始信息汇总
FoldBench数据集概述
数据集简介
- 名称: FoldBench: An All-atom Benchmark for Biomolecular Structure Prediction
- 类型: 生物分子结构预测基准测试集
- 特点: 低同源性,涵盖蛋白质、核酸、配体及六种主要相互作用类型
评估内容
蛋白质相互作用
- 评估指标: 成功率(Success Rate)
- 评估模型:
- AlphaFold 3
- Boltz-1
- Chai-1
- HelixFold 3
- Protenix
- 评估子类:
- Protein-Protein
- Antibody–Antigen
- Protein-Ligand
核酸相互作用
- 评估指标:
- 界面预测:成功率(Success Rate)
- 单体预测:LDDT(Local Distance Difference Test)
- 评估模型:
- AlphaFold 3
- Boltz-1
- Chai-1
- HelixFold 3
- Protenix
- 评估子类:
- Protein-RNA
- Protein-DNA
- RNA Monomer
- DNA Monomer
成功标准
- Protein–Ligand界面: LRMSD < 2 Å 且 LDDT-PLI > 0.8
- 其他界面: DockQ ≥ 0.23
引用信息
bibtex @article {Xu2025.05.22.655600, author = {Xu, Sheng and Feng, Qiantai and Qiao, Lifeng and Wu, Hao and Shen, Tao and Cheng, Yu and Zheng, Shuangjia and Sun, Siqi}, title = {FoldBench: An All-atom Benchmark for Biomolecular Structure Prediction}, year = {2025}, doi = {10.1101/2025.05.22.655600}, journal = {bioRxiv} }
致谢
数据集开发感谢以下项目的贡献者:
- Alphafold3
- Protenix
- Chai-1
- Boltz-1
- Helixfold3
- Openstructure
- DockQ
搜集汇总
数据集介绍
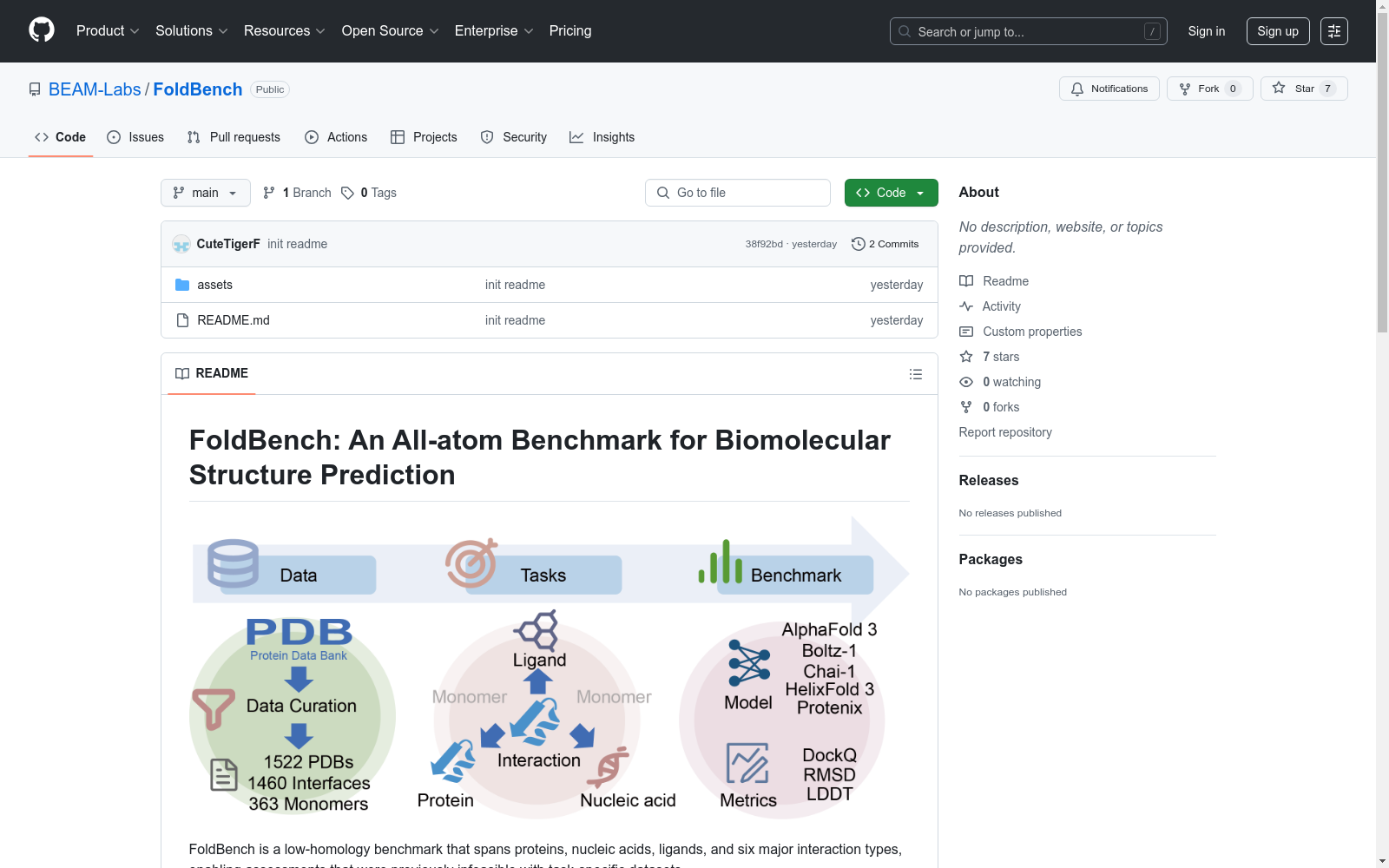
构建方式
FoldBench作为生物分子结构预测领域的重要基准数据集,其构建过程体现了严谨的科学方法论。研究团队采用低同源性的筛选策略,精心选取涵盖蛋白质、核酸、配体等生物分子的结构数据,并整合六种主要相互作用类型。通过多维度评估指标(如LRMSD、LDDT-PLI和DockQ)建立标准化评价体系,确保数据集的全面性和代表性。该数据集特别关注分子界面预测的准确性,为复杂生物分子系统的结构预测研究提供了可靠基准。
使用方法
使用FoldBench进行模型评估时,研究者需遵循其标准化的测试协议。对于蛋白质相互作用预测,应采用DockQ≥0.23作为成功阈值;而蛋白质-配体界面预测则需同时满足LRMSD<2Å和LDDT-PLI>0.8的双重标准。数据集提供的详细性能排行榜(如AlphaFold3、Boltz-1等主流模型的对比数据)可作为算法优化的参照系。通过调用官方提供的评估脚本,用户可自动计算各项指标并生成标准化报告,确保结果的可比性和可重复性。
背景与挑战
背景概述
FoldBench是由Sheng Xu等研究人员于2025年提出的一个全原子生物分子结构预测基准数据集,旨在解决生物分子结构预测领域中的多类型相互作用评估问题。该数据集涵盖了蛋白质、核酸、配体及其六种主要相互作用类型,通过低同源性的设计策略,填补了传统任务特定数据集无法全面评估跨分子类型预测性能的空白。FoldBench的建立为AlphaFold 3、Boltz-1等前沿模型提供了标准化测试平台,其创新性的接口成功率和LDDT评估指标,显著推动了生物分子结构预测领域的方法开发和性能比较研究。
当前挑战
FoldBench面临的挑战主要体现在两个方面:在领域问题层面,生物分子结构预测涉及复杂的多尺度相互作用,特别是蛋白质-核酸、抗体-抗原等界面的精确建模存在显著难度,现有模型在跨类型相互作用预测中表现差异较大;在构建过程层面,低同源性样本的筛选需要平衡结构多样性与可计算性,而全原子级别的评估指标设计需兼顾不同分子类型的物理化学特性,这对数据集的代表性和可解释性提出了极高要求。
常用场景
经典使用场景
FoldBench作为生物分子结构预测领域的全原子基准测试集,其经典使用场景在于评估蛋白质、核酸、配体及其相互作用预测模型的性能。该数据集通过低同源性的设计,有效避免了模型因数据偏差而导致的过拟合问题,为研究者提供了全面、客观的评估平台。尤其在蛋白质-蛋白质、抗体-抗原、蛋白质-配体等复杂相互作用场景中,FoldBench能够精准衡量模型的预测精度和泛化能力。
解决学术问题
FoldBench解决了生物分子结构预测领域中长期存在的评估标准不统一、数据覆盖不全等关键学术问题。传统数据集往往局限于单一分子类型或相互作用,难以全面反映模型的真实性能。FoldBench通过整合六种主要相互作用类型,填补了跨分子类别评估的空白,为研究者提供了更全面的性能分析工具。其严格的评估指标(如LRMSD和DockQ)进一步提升了结果的可比性和科学性。
实际应用
在实际应用中,FoldBench为药物发现、蛋白质设计等生物技术领域提供了重要支持。通过评估不同模型在蛋白质-配体相互作用预测中的表现,研究人员能够筛选出最适合特定药物靶点识别的算法。同时,该数据集在核酸结构预测方面的表现评估,为基因治疗和合成生物学中的分子设计提供了可靠参考。FoldBench的广泛应用显著加速了生物分子工程领域的研发进程。
数据集最近研究
最新研究方向
FoldBench作为生物分子结构预测领域的全原子基准测试集,近期研究聚焦于多模态分子相互作用的高精度建模。随着AlphaFold3等深度学习模型在蛋白质单体预测取得突破后,学术界正将注意力转向蛋白质-核酸、蛋白质-配体等复合体界面预测的泛化能力评估。该数据集通过整合六类相互作用类型和低同源序列,为跨分子类别的三维结构预测模型提供了系统性验证平台。当前热点集中在利用几何约束和物理启发的神经网络架构,提升模型在抗体-抗原结合位点等挑战性场景的预测鲁棒性。FoldBench的评估框架正推动着从单一分子向生物分子网络的功能性结构预测范式转变。
以上内容由遇见数据集搜集并总结生成


