Some <i>ab initio</i> thoughts on the bonding in O<sub>3</sub>H
收藏DataCite Commons2021-03-22 更新2024-08-17 收录
下载链接:
https://tandf.figshare.com/articles/dataset/Some_i_ab_initio_i_thoughts_on_the_bonding_in_O_sub_3_sub_H/12859744
下载链接
链接失效反馈官方服务:
资源简介:
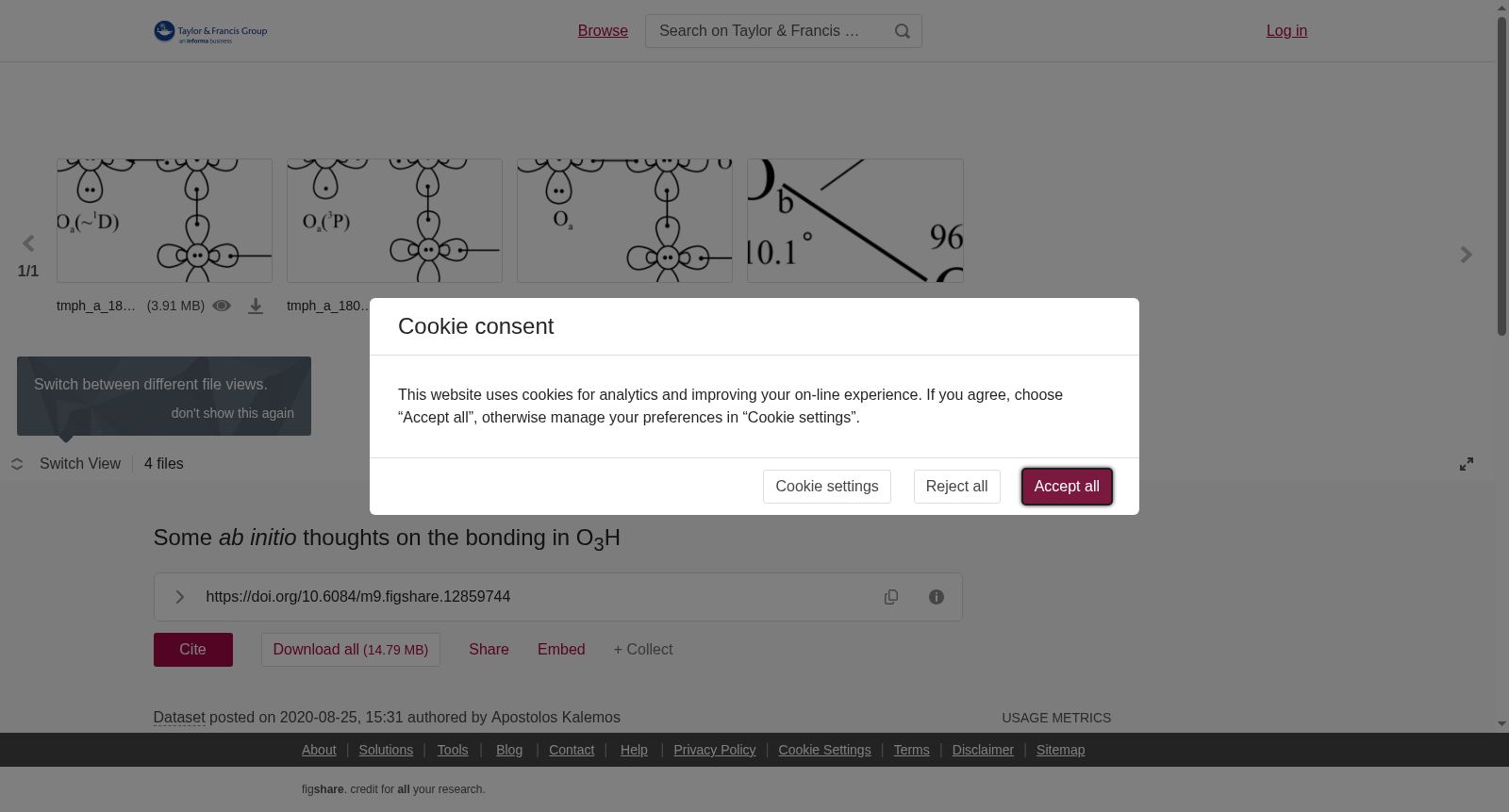
We study the ground X~2A′′ state of O<sub>3</sub>H (= O<sub>a</sub>O<sub>b</sub>O<sub>c</sub>H) with single (RCCSD(T)) and multi (MRCI) reference correlation methods in order to shed some light on its bonding mechanism in connection with its low dissociation energy and rather long bond distance (O<sub>a</sub>O<sub>b</sub>−O<sub>c</sub>H). For such a task all three dissociation/formation paths were considered (O<sub>2 </sub>+ OH, O + O<sub>2</sub>H, and O<sub>3 </sub>+ H) and the associated nonadiabatic coupling matrix elements were examined. It appears that the excited states of the above asymptotic fragments participate in the equilibrium wavefunction of O<sub>3</sub>H in a way that results in a symmetry broken structure.
本研究采用单参考(RCCSD(T))与多参考(MRCI)相关电子结构方法,对O₃H(即OₐOᵦO꜀H)的基态X~2A''态开展理论探究,以期阐明其成键机制——该机制与其低解离能及较长的OₐOᵦ−O꜀H键长密切相关。为完成此项研究,我们考量了全部三条解离/形成路径:O₂ + OH、O + O₂H与O₃ + H,并对与之相关的非绝热耦合矩阵元进行了考察。研究结果表明,上述渐近解离碎片的激发态以特定方式参与O₃H的平衡波函数,最终使体系呈现对称性破缺结构。
提供机构:
Taylor & Francis创建时间:
2020-08-25
搜集汇总
数据集介绍
背景与挑战
背景概述
该数据集聚焦于O3H分子的键合机制研究,采用单参考和多参考相关方法分析其基态特性,重点探讨低解离能和长键距的成因。研究通过三种解离路径和非绝热耦合元素分析,揭示激发态参与导致对称性破缺结构,为理解O3H的化学键行为提供理论依据。
以上内容由遇见数据集搜集并总结生成


