graphs-datasets/MD17-aspirin
收藏Hugging Face2023-02-07 更新2024-03-04 收录
下载链接:
https://hf-mirror.com/datasets/graphs-datasets/MD17-aspirin
下载链接
链接失效反馈官方服务:
资源简介:
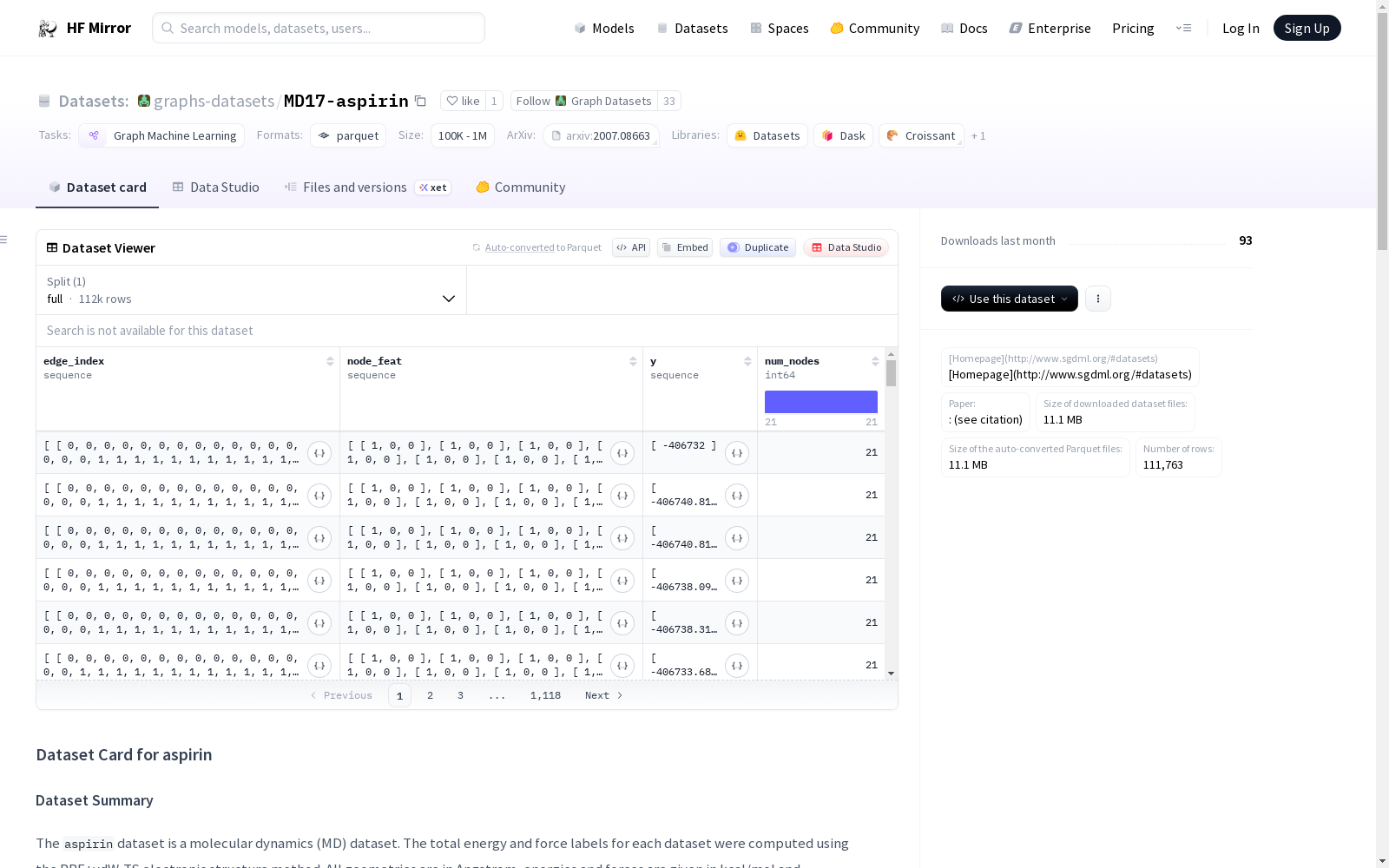
`aspirin`数据集是一个分子动力学(MD)数据集,使用PBE+vdW-TS电子结构方法计算每个数据的总能量和力标签。所有几何结构以埃为单位,能量和力分别以kcal/mol和kcal/mol/A为单位。该数据集主要用于有机分子属性预测,是一个回归任务,涉及1个属性的预测。数据集的规模较大,包含111762个图,平均每个图有21个节点和303.0447106824262条边。
The `aspirin` dataset is a molecular dynamics (MD) dataset, where the total energy and force labels for each entry are calculated using the PBE+vdW-TS electronic structure method. All geometric structures are reported in angstroms, while energy and force values are measured in kcal/mol and kcal/mol/Å, respectively. This dataset is primarily used for organic molecular property prediction, serving as a regression task that involves predicting a single property. The dataset has a large scale, containing 111,762 molecular graphs, with an average of 21 nodes and 303.0447106824262 edges per graph.
提供机构:
graphs-datasets
原始信息汇总
数据集概述
数据集描述
- 数据集名称: aspirin
- 数据集类型: 分子动力学(MD)数据集
- 计算方法: 使用PBE+vdW-TS电子结构方法计算总能量和力标签
- 单位: 几何结构单位为Angstrom,能量单位为kcal/mol,力单位为kcal/mol/A
支持的任务和排行榜
- 任务类型: 有机分子属性预测
- 任务性质: 回归任务
- 预测属性数量: 1
- 评分标准: 能量预测的平均绝对误差(meV)
数据集结构
-
数据属性
- 规模: 大
- 图数量: 111762
- 平均节点数: 21.0
- 平均边数: 303.0447106824262
-
数据字段
node_feat(列表: #nodes x #node-features): 节点特征edge_index(列表: 2 x #edges): 构成边的节点对edge_attr(列表: #edges x #edge-features): 边特征y(列表: #labels): 可用于预测的标签数量num_nodes(整数): 图的节点数
-
数据分割: 未分割,建议使用交叉验证
附加信息
- 许可证信息: 未知
- 引用信息: 参见提供的引用文献
外部使用
- PyGeometric: 使用PyGeometric加载数据集的示例代码提供
以上概述基于提供的数据集详情页面内容,确保了信息的准确性和相关性。
搜集汇总
数据集介绍
构建方式
在计算化学领域,分子动力学模拟为理解分子行为提供了关键数据。MD17-aspirin数据集的构建源于对阿司匹林分子高精度模拟的需求,通过采用PBE+vdW-TS电子结构方法,系统计算了每个构型的总能量与原子受力。所有几何结构以埃为单位记录,能量与力的单位分别为千卡每摩尔与千卡每摩尔每埃,确保了数据的物理一致性。该数据集包含111,762个分子构型,每个构型平均包含21个原子节点与303条边,全面覆盖了阿司匹林分子的动态行为。
使用方法
为有效利用该数据集,研究者可借助HuggingFace的datasets库直接加载,并通过PyGeometric转换为图数据对象。典型流程包括使用DataLoader构建迭代器,以支持批量训练。在任务设计上,应聚焦于能量预测的回归问题,采用平均绝对误差作为评估指标。由于数据未预先分割,建议实施交叉验证策略,以确保模型泛化能力。数据字段涵盖节点特征、边索引、边属性及标签,为构建端到端的分子性质预测模型提供了完整输入。
背景与挑战
背景概述
在计算化学与分子动力学领域,精确预测分子系统的能量与力场是模拟物质行为的关键基础。MD17-aspirin数据集作为分子动力学基准数据,由Stefan Chmiela等研究人员于2017年构建,依托PBE+vdW-TS电子结构方法计算阿司匹林分子的总能量与原子间作用力。该数据集收录了超过11万分子构型,每个构型包含21个原子节点及丰富的边特征,旨在推动机器学习模型在有机分子性质预测任务中的应用,为开发高精度、高效率的力场模型提供了重要数据支撑,显著促进了计算化学与人工智能的交叉融合。
当前挑战
该数据集致力于解决分子动力学中力场构建的挑战,即如何通过机器学习方法从量子化学计算数据中学习出既精确又高效的原子间势能函数,以替代传统计算成本高昂的从头算方法。在构建过程中,研究人员面临多重挑战:首先,需确保数据的高精度与一致性,所有几何结构、能量及力标签均需基于可靠的PBE+vdW-TS方法生成;其次,数据规模庞大且结构复杂,涉及大量分子构型与高维特征,对存储、处理及模型训练提出了较高要求;此外,数据未预设标准划分,需依赖交叉验证策略进行评估,增加了模型泛化能力验证的复杂度。
常用场景
经典使用场景
在计算化学与分子动力学领域,MD17-aspirin数据集作为经典基准,广泛用于有机分子性质预测任务。该数据集包含阿司匹林分子的高精度能量与力场标签,通过图神经网络模型,研究者能够学习分子结构的图表示,进而回归预测其总能量与原子间作用力。这一场景常被用于评估模型在量子化学计算中的准确性,为分子模拟提供高效的数据驱动方法。
解决学术问题
MD17-aspirin数据集有效解决了传统分子动力学模拟中计算成本高昂的学术难题。通过提供基于PBE+vdW-TS电子结构方法生成的精确能量与力标签,该数据集使得机器学习模型能够学习并逼近复杂的量子力学势能面。这不仅加速了分子性质预测的过程,还促进了能量守恒力场的发展,为计算化学领域提供了可扩展且高精度的研究工具。
实际应用
在实际应用中,MD17-aspirin数据集被用于药物设计与材料科学中的分子模拟优化。基于该数据集训练的模型能够快速预测阿司匹林等有机分子的稳定构象与反应路径,辅助研究人员筛选候选药物或分析分子相互作用。这种数据驱动方法显著降低了实验与计算成本,为工业界的分子工程与生物化学研究提供了可靠支持。
数据集最近研究
最新研究方向
在计算化学与材料科学领域,分子动力学数据集如MD17-aspirin正推动着机器学习与量子力学计算的深度融合。该数据集以其高精度的能量与力标签,成为开发守恒性分子力场模型的关键基准。前沿研究聚焦于利用图神经网络架构,如消息传递机制,以原子间相互作用为边特征,精准预测分子系统的势能面。这一方向不仅加速了新材料的虚拟筛选进程,更在药物分子构象模拟中展现出巨大潜力,为降低传统密度泛函理论计算的高昂成本提供了可行路径。
以上内容由遇见数据集搜集并总结生成


