FAPbI3_tetragonal&cubic
收藏Mendeley Data2024-04-30 更新2024-06-27 收录
下载链接:
https://www.scidb.cn/en/detail?dataSetId=104766369689486fb5039c19be341b88
下载链接
链接失效反馈官方服务:
资源简介:
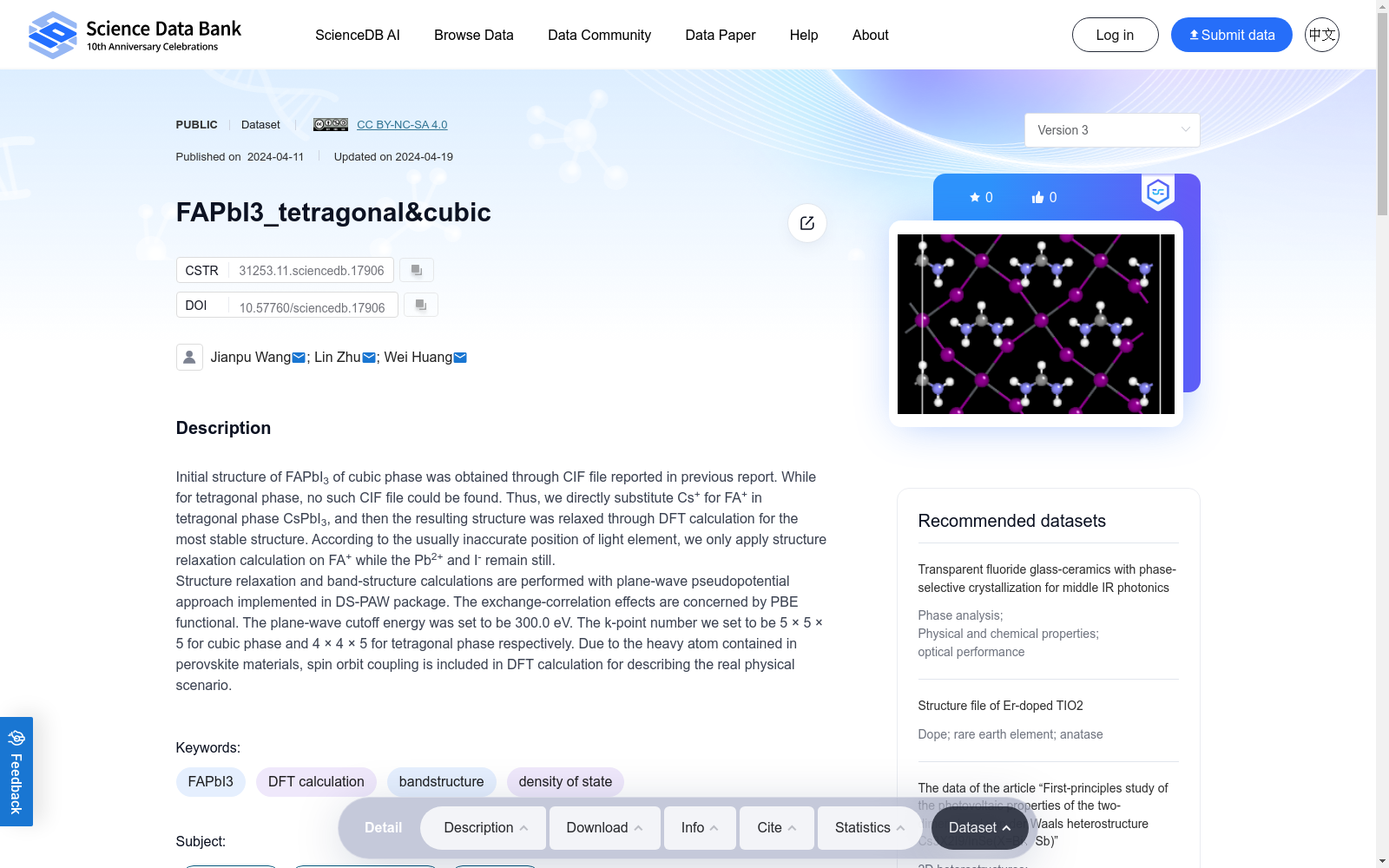
Initial structure of FAPbI3 of cubic phase was obtained through CIF file reported in previous report. While for tetragonal phase, no such CIF file could be found. Thus, we directly substitute Cs+ for FA+ in tetragonal phase CsPbI3, and then the resulting structure was relaxed through DFT calculation for the most stable structure. According to the usually inaccurate position of light element, we only apply structure relaxation calculation on FA+ while the Pb2+ and I- remain still.Structure relaxation and band-structure calculations are performed with plane-wave pseudopotential approach implemented in DS-PAW package. The exchange-correlation effects are concerned by PBE functional. The plane-wave cutoff energy was set to be 300.0 eV. The k-point number we set to be 5 × 5 × 5 for cubic phase and 4 × 4 × 5 for tetragonal phase respectively. Due to the heavy atom contained in perovskite materials, spin orbit coupling is included in DFT calculation for describing the real physical scenario.
立方相FAPbI3的初始结构通过既往文献报道的CIF(晶体学信息文件,Crystallographic Information File)文件获取。针对四方相FAPbI3,目前未发现可用的CIF文件,因此我们以四方相CsPbI3为模板,将其中的FA+替换为Cs+,随后通过DFT(密度泛函理论,Density Functional Theory)计算对所得结构进行弛豫以得到最稳定构型。考虑到轻元素的原子位置通常存在较大误差,我们仅对FA+进行结构弛豫计算,而保持Pb²+与I⁻的位置固定。结构弛豫与能带结构计算采用DS-PAW软件包所实现的平面波赝势方法完成。交换关联效应通过PBE(Perdew–Burke–Ernzerhof)泛函进行描述,平面波截断能量设置为300.0 eV。我们分别为立方相与四方相设置了5×5×5与4×4×5的k点网格。鉴于钙钛矿材料中含有重原子,我们在DFT计算中引入自旋轨道耦合以更准确地还原真实物理场景。
创建时间:
2024-04-14
搜集汇总
数据集介绍
背景与挑战
背景概述
该数据集提供了钙钛矿材料FAPbI3的四方相和立方相结构数据,其中立方相基于已有CIF文件,四方相通过DFT计算从CsPbI3结构替换并松弛获得。数据集包含通过DS-PAW软件包进行的密度泛函理论计算,涉及结构松弛和能带结构分析,并考虑了自旋轨道耦合效应,适用于材料科学和物理化学研究。
以上内容由遇见数据集搜集并总结生成


