FAPbI3_tetragonal&cubic
收藏Mendeley Data2024-04-20 更新2024-06-27 收录
下载链接:
https://www.doi.org/10.57760/sciencedb.17906
下载链接
链接失效反馈官方服务:
资源简介:
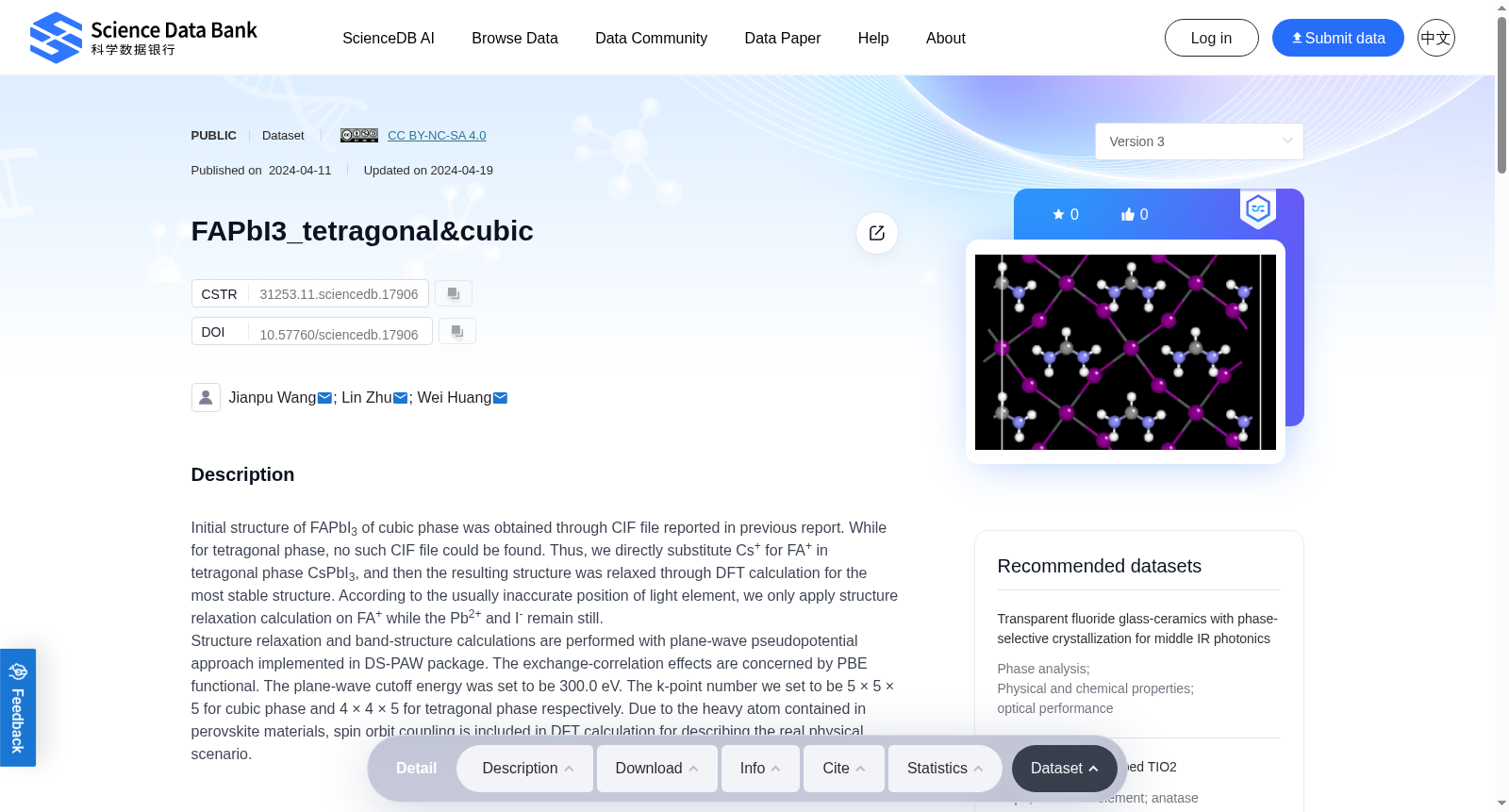
Initial structure of FAPbI3 of cubic phase was obtained through CIF file reported in previous report. While for tetragonal phase, no such CIF file could be found. Thus, we directly substitute Cs+ for FA+ in tetragonal phase CsPbI3, and then the resulting structure was relaxed through DFT calculation for the most stable structure. According to the usually inaccurate position of light element, we only apply structure relaxation calculation on FA+ while the Pb2+ and I- remain still.Structure relaxation and band-structure calculations are performed with plane-wave pseudopotential approach implemented in DS-PAW package. The exchange-correlation effects are concerned by PBE functional. The plane-wave cutoff energy was set to be 300.0 eV. The k-point number we set to be 5 × 5 × 5 for cubic phase and 4 × 4 × 5 for tetragonal phase respectively. Due to the heavy atom contained in perovskite materials, spin orbit coupling is included in DFT calculation for describing the real physical scenario.
创建时间:
2024-04-20
搜集汇总
数据集介绍
背景与挑战
背景概述
该数据集提供了FAPbI3立方相和四方相的初始结构及其DFT计算结果,包括能带结构和态密度,适用于材料科学和物理学研究。
以上内容由遇见数据集搜集并总结生成


