variant-benchmark
收藏Hugging Face2025-04-02 更新2025-04-02 收录
下载链接:
https://huggingface.co/datasets/m42-health/variant-benchmark
下载链接
链接失效反馈官方服务:
资源简介:
variant-benchmark数据集是一个基因组学基准测试,用于评估模型在不同生物学背景下利用变异信息的有效性。它包含了编码和非编码区域的致病性评估、基因表达效应预测、剪接变异效应评估、DNA甲基化影响评估、祖先分类以及常见与合成变异的区分等多个生物信息学任务。
创建时间:
2025-03-26
搜集汇总
数据集介绍
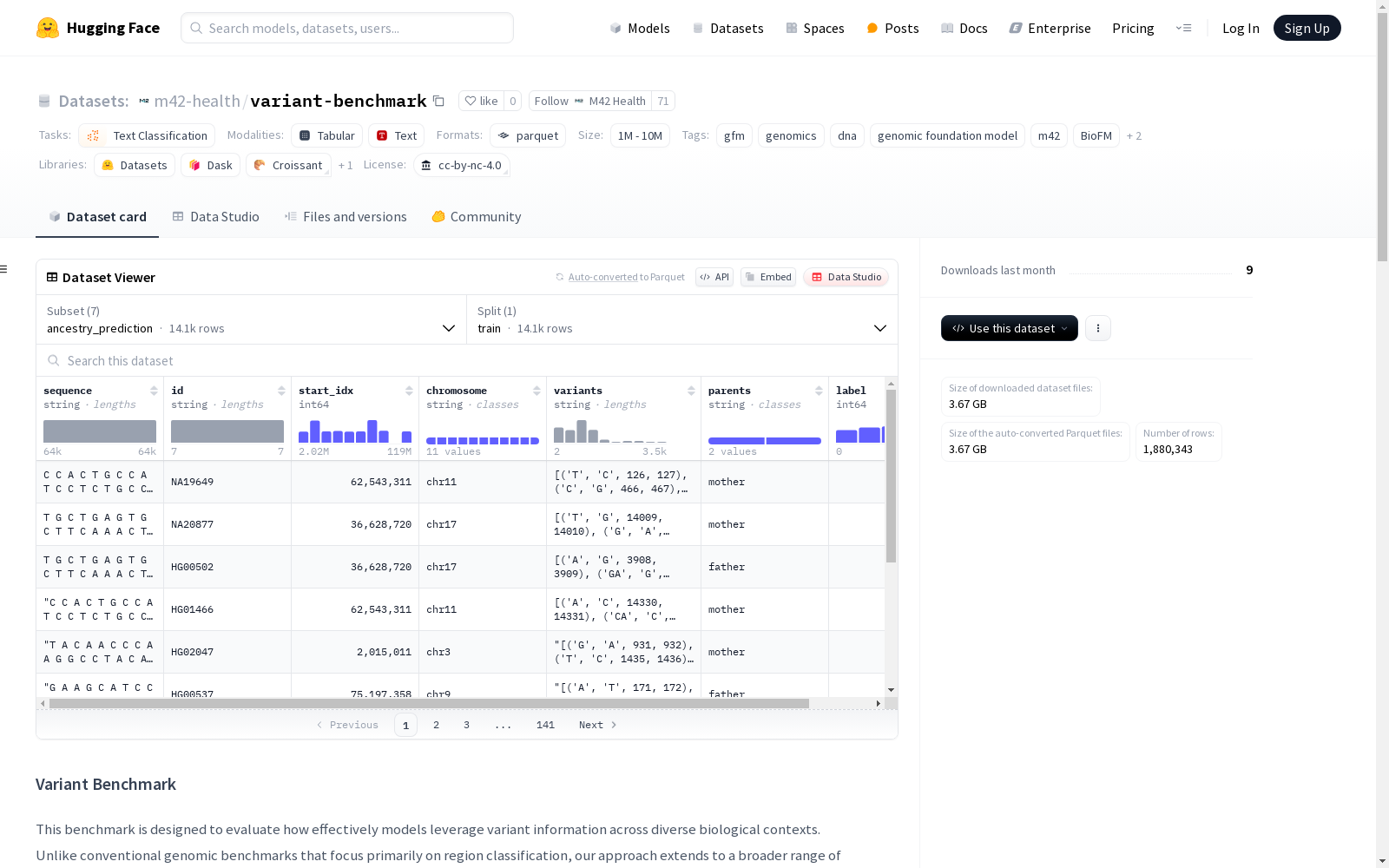
构建方式
variant-benchmark数据集通过整合多个权威生物信息学资源构建而成,涵盖七种关键基因组学任务。在编码区致病性评估中采用AlphaMissense数据库的变异注释,非编码区致病性分析则基于BEND数据集的29.5万个单核苷酸变异。表达效应预测整合了DeepSea的调控注释数据,而剪接数量性状位点(sQTL)分析则源自包含百万级变异-组织对的sqtlSeeker2数据集。甲基化定量性状位点(meQTL)任务采用了GRASP数据库的变异-甲基化关联数据,群体结构分类则基于千人基因组计划的五大超群体标记数据。
特点
该数据集最显著的特点是实现了多维度基因组功能评估的系统性整合。其包含超过105万条变异-组织对的剪接数据,29.5万非编码变异的致病性标注,以及15.6万变异相关的表达调控注释。每个子集均提供染色体定位、序列上下文、等位基因信息等结构化特征,其中表达预测任务还包含DeepSea的多标签注释。数据集采用标准化的变异表示方法,统一包含参考序列和变异序列的侧翼上下文信息,确保不同任务间数据格式的一致性。
使用方法
使用该数据集时,可通过Hugging Face的datasets库按需加载特定子集。加载时需指定七类任务之一作为subset参数,包括ancestry_prediction、coding_pathogenicity等。所有子集默认包含训练分割,建议采用k折交叉验证进行模型评估。数据加载后以字典形式返回,包含序列特征、染色体位置、变异信息和标签等字段。例如表达预测任务返回alt_left/right和ref_left/right等序列上下文特征,以及deepsea_label等多标签注释,便于进行基因组功能预测任务的模型训练与评估。
背景与挑战
背景概述
variant-benchmark数据集由M42 Health团队于2025年创建,旨在评估基因组基础模型在多种生物背景下利用变异信息的有效性。该数据集整合了来自AlphaMissense、BEND、DeepSea等多个权威来源的基因组数据,涵盖了编码区致病性预测、非编码区致病性评估、基因表达效应预测等七项核心任务。作为基因组基础模型领域的重要评估工具,该数据集突破了传统基因组基准测试的局限,首次实现了对变异介导的多重分子机制的综合性评估,为精准医学和功能基因组学研究提供了新的方法论支持。
当前挑战
variant-benchmark数据集面临双重挑战:在科学层面,需要解决基因组变异跨机制功能预测的复杂性,特别是非编码区变异与基因调控网络相互作用的建模难题;在技术层面,数据集构建涉及多源异构数据整合,包括来自1000 Genomes Project的群体遗传数据与sqtlSeeker2的剪接定量性状位点数据,需克服数据格式标准化、样本平衡性控制等技术障碍。此外,合成控制变体的生成策略需确保其与真实常见变异的生物学背景可比性,这对基准测试的可靠性提出了更高要求。
常用场景
经典使用场景
在基因组学研究领域,variant-benchmark数据集通过整合多种变异驱动的分子过程评估任务,为功能基因组学模型提供了全面的性能验证平台。该数据集特别适用于评估模型在编码区/非编码区致病性预测、基因表达调控、可变剪接效应等核心生物机制中的表现,其多任务架构设计显著提升了模型在跨场景应用中的泛化能力。
实际应用
在临床基因组学实践中,该数据集支撑的模型评估可直接转化为精准医疗应用。基于编码区致病性预测模块优化的算法已用于遗传病诊断系统;sQTL任务驱动的剪接变异分析为癌症基因组学研究提供工具;而甲基化定量性状位点(meQTL)评估则助力复杂疾病风险预测模型的开发。
衍生相关工作
该数据集催生了BioToken等生物信息学基础模型的创新研究,其多任务评估框架被后续工作如GenomicBERT、DNABERT-2广泛采用。基于该基准开发的跨模态注意力机制,在《Nature Machine Intelligence》等期刊发表的系列研究中,显著提升了变异功能预测的准确度与可解释性。
以上内容由遇见数据集搜集并总结生成


