depinwang/rnaseq-aligner-toy-benchmark-metrics-v2-starsweep
收藏Hugging Face2026-05-01 更新2026-05-03 收录
下载链接:
https://hf-mirror.com/datasets/depinwang/rnaseq-aligner-toy-benchmark-metrics-v2-starsweep
下载链接
链接失效反馈官方服务:
资源简介:
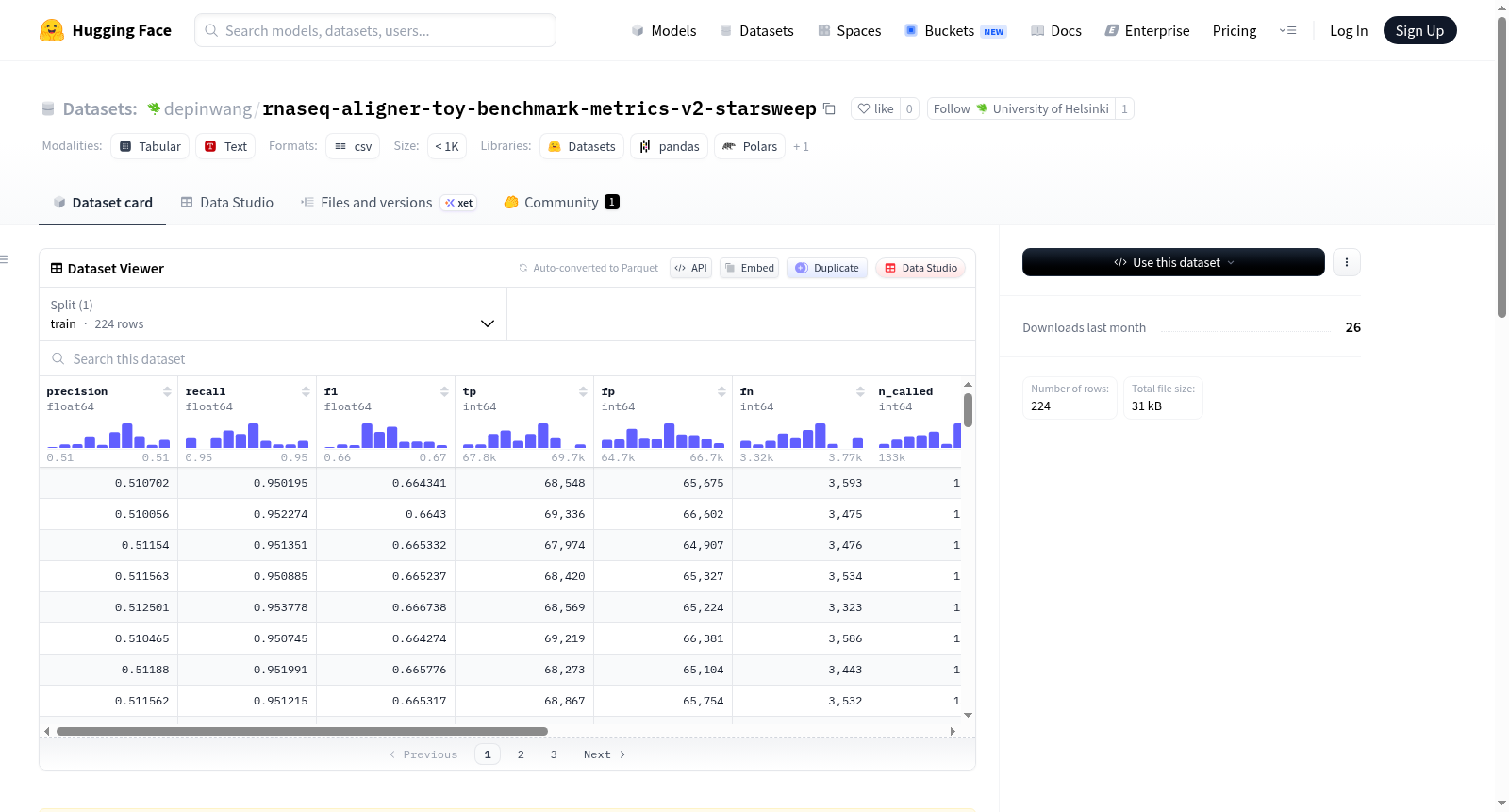
该数据集名为RNA-seq Aligner Benchmark — Metrics (v2-starsweep),包含了模拟GRCh38 RNA-seq reads与GENCODE v45转录本的真实内含子之间的比对结果。具体来说,它记录了每个(比对工具,样本)组合的剪接连接点F1分数、精确率和召回率等性能指标。数据集共有224行,每行包含比对工具名称(如star、hisat2、subjunc、bwa-mem2、minimap2)、样本ID、数据集标识符、精确率、召回率、F1分数、真阳性、假阳性、假阴性、调用的连接点总数、真实连接点总数以及应用的读支持过滤器等信息。数据集的来源信息显示,实验是在Puhti (CSC)集群上进行的,参考基因组为GRCh38.primary_assembly + GENCODE v45,模拟方法为simulate_reads.py(均匀采样转录本,无错误模型),评分方法为score_junctions.py(精确元组匹配,min_reads=3)。
The dataset is named RNA-seq Aligner Benchmark — Metrics (v2-starsweep) and contains the splice-junction F1 / precision / recall metrics for simulated GRCh38 RNA-seq reads aligned against ground-truth introns from GENCODE v45 transcripts. Specifically, it records the performance metrics for each (aligner, sample) combination, including aligner name (e.g., star, hisat2, subjunc, bwa-mem2, minimap2), sample ID, dataset identifier, precision, recall, f1 score, true positives, false positives, false negatives, total junctions called, total junctions in ground-truth set, and the read-support filter applied. The dataset consists of 224 rows. Provenance information indicates that the experiment was conducted on the Puhti (CSC) cluster, using the GRCh38.primary_assembly + GENCODE v45 reference, with reads simulated using simulate_reads.py (uniformly sampled transcripts, no error model) and scored using score_junctions.py (exact tuple match, min_reads=3).
提供机构:
depinwang
搜集汇总
数据集介绍
构建方式
本数据集源于对五种经典RNA-seq比对工具(STAR、HISAT2、Subjunc、BWA-MEM2及Minimap2)在剪接连接点识别性能上的系统性评估。借助GENCODE v45注释中的真实内含子信息,基于GRCh38参考基因组,通过无错误模型的转录本均匀采样生成模拟读段。每个(比对器,样本)组合均经严格评分流程处理,以精确匹配元组的方式计算混淆矩阵指标,并设定最小读段支持阈值为3,最终汇集而成224行结构化的基准测试指标。
使用方法
用户可直接读取CSV格式的表格,利用'aligner'、'sample'与'dataset'字段进行分组聚合或筛选。借助'precision'、'recall'与'f1'列可快速评估各比对器的整体性能排序;而'tp'、'fp'与'fn'则适合进行更为细致的统计检验或误差分析。此外,'min_reads'列可用于模拟不同严格度过滤条件对结果的影响,灵活适配不同研究场景对比工具效用的需求。
背景与挑战
背景概述
RNA测序数据分析中,剪接位点的准确识别是解析基因表达与可变剪接调控机制的核心环节。然而,不同比对算法在处理模拟或真实测序数据时,其剪接位点检测性能存在显著差异,亟需标准化评估基准。rnaseq-aligner-toy-benchmark-metrics-v2-starsweep数据集由CSC的Puhti集群于近期创建,旨在系统评估STAR、HISAT2、Subjunc、BWA-MEM2及Minimap2等五种主流比对器在模拟GRCh38 RNA-seq读段上的剪接位点检测效能。该数据集以GENCODE v45转录本的真实内含子为金标准,通过精确率、召回率与F1分数等指标量化比对器性能,为RNA-seq分析流程的算法选择与优化提供了可靠参考基准,对推动转录组学研究的可重复性具有重要影响。
当前挑战
该数据集所应对的核心领域挑战在于缺乏统一、精细的比对器性能评估标准,尤其在剪接位点检测层面,不同工具对复杂可变剪接事件的敏感性与特异性差异显著,直接影响下游差异表达与剪接功能解析的可靠性。构建过程中,挑战主要体现在三个方面:一是模拟数据的真实性平衡,采用无错误模型的均匀采样方式可能无法全面反映真实测序噪声与转录本丰度偏差;二是金标准定义需依赖GENCODE v45注释的完整性,但注释中罕见或新发现剪接事件可能缺失,导致假阴性被高估;三是评价参数如最小读段支持阈值(min_reads=3)的设定虽可减少噪声,但会偏向高表达剪接位点,削弱对低丰度事件的检测能力评估。
常用场景
经典使用场景
在转录组学研究中,RNA-seq比对器的性能评估是确保下游差异表达和可变剪接分析准确性的关键环节。该数据集专为系统评估五种主流比对器(STAR、HISAT2、Subjunc、BWA-MEM2、Minimap2)而设计,通过模拟GRCh38参考基因组上的RNA-seq读段,并以GENCODE v45注释中的真实内含子为金标准,计算剪接连接点的精确率、召回率和F1分数。其经典使用场景为在标准化流程下横向对比不同比对器的剪接检测能力,研究者可据此筛选最适合自身实验设计的工具,或优化比对参数以提升特定样本类型的定量可靠性。
解决学术问题
该数据集直面RNA-seq比对领域长期存在的基准缺失问题,即缺乏一个统一、可复现的指标框架来客观衡量比对器在剪接连接点层面的真实性能。通过提供精确率、召回率和F1分数等细粒度指标,它解决了如何量化比对器对真实剪接事件的捕捉能力这一核心学术难题,尤其有助于揭示比对器在低表达剪接位点或复杂结构变异上的系统偏差。其意义在于推动了比对算法的标准化评估,使研究者能够摒弃主观偏好,基于实证数据选择工具,从而显著提升可变剪接研究的可重复性和结论的稳健性。
实际应用
在转化医学和临床RNA-seq数据分析的实际工作中,该数据集的应用场景极具针对性。例如,癌症基因组学中需要精准识别肿瘤特异性融合基因或异常剪接亚型,研究人员可借助该基准数据快速筛选出对罕见剪接事件召回率最高的比对器,避免因工具选择不当而遗漏关键生物标志物。此外,在药物基因组学中,评估药物靶点基因的剪接变体表达量时,该数据集提供的性能指标能帮助优化分析流程,确保后续定量结果的临床决策支持价值。同时,它也为生物信息学工具开发者提供了调试和验证比对算法性能的关键参考。
数据集最近研究
最新研究方向
该数据集聚焦于RNA-seq比对工具在模拟读段上的剪接连接点检测性能评估,通过精确率、召回率和F1分数等指标,系统比较STAR、HISAT2等主流比对器在GRCh38参考基因组与GENCODE v45真实内含子注释下的表现。最新研究趋势正从单一比对精度转向多维度评估,包括低表达剪接事件检测、跨平台稳健性以及基于深度学习的校正方法。与之关联的热点事件包括ENCODE等大规模测序项目对剪接注释的标准化需求,以及个性化医疗中肿瘤特异性融合基因发现的精度挑战。此基准为优化比对算法、确保转录组分析可靠性提供关键基准,尤其推动长读长测序时代下剪接变异的精准识别,对疾病机制解析与生物标志物开发具有基础支撑价值。
以上内容由遇见数据集搜集并总结生成


