geom_drugs
收藏Hugging Face2026-04-30 更新2026-05-01 收录
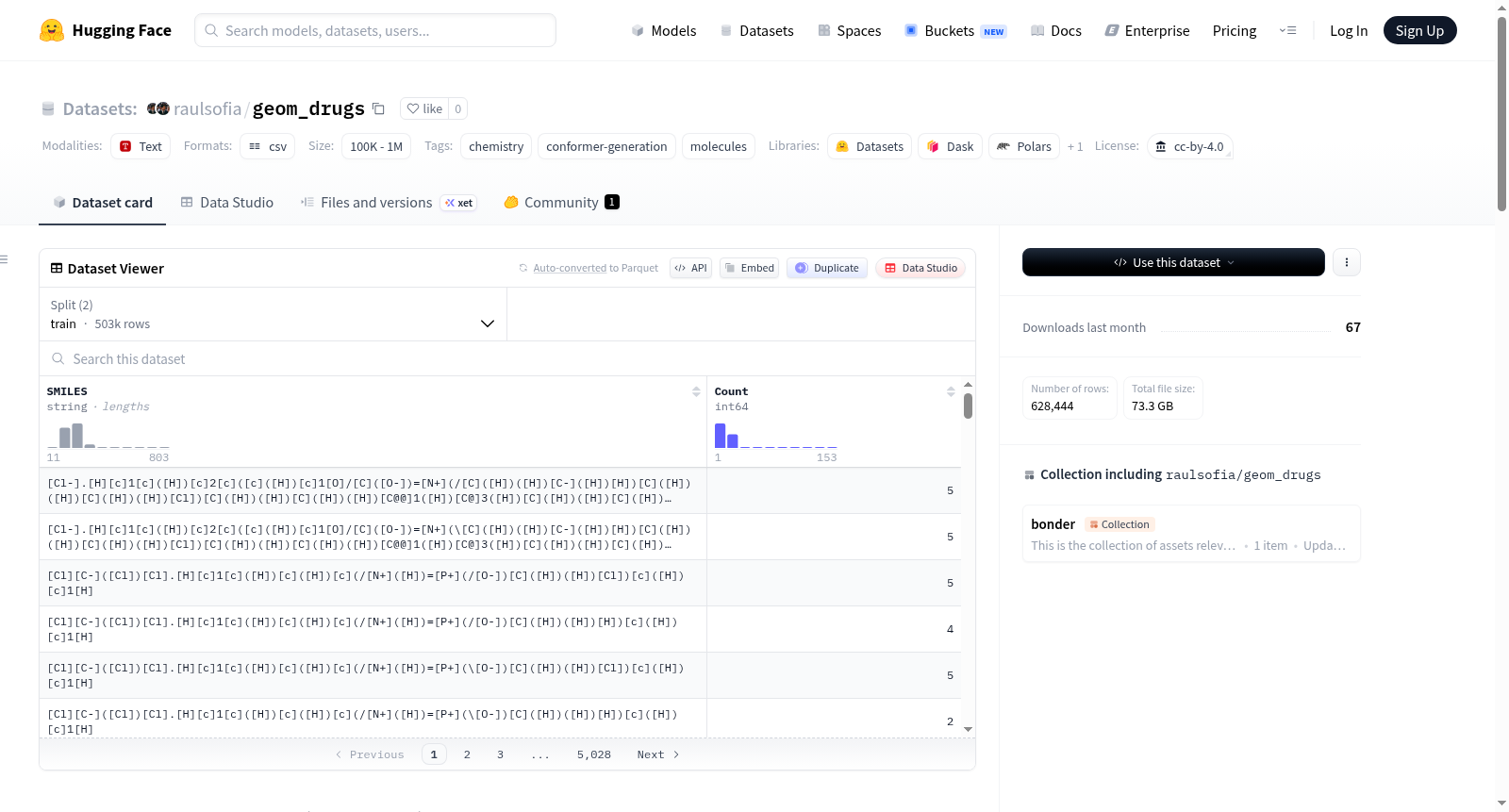
下载链接:
https://huggingface.co/datasets/raulsofia/geom_drugs
下载链接
链接失效反馈官方服务:
资源简介:
GEOM-Drugs数据集是GEOM数据集(药物子集)的一个镜像和预处理版本,专注于具有实验数据的药物样分子。该数据集包含数百万个经过能量和统计权重注释的分子构象。原始数据集由Simon Axelrod和Rafael Gómez-Bombarelli创建,用于属性预测和分子生成。此版本通过Git分支管理项目特定的预处理步骤和数据分割策略,确保数据的可重现性和项目隔离。使用该数据集时需引用原始论文。
The GEOM-Drugs dataset is a mirrored and preprocessed version of the GEOM dataset (drug subset), focusing on drug-like molecules with experimental data. This dataset contains millions of molecular conformations annotated with energy and statistical weights. The original dataset was created by Simon Axelrod and Rafael Gómez-Bombarelli for property prediction and molecular generation. This version manages project-specific preprocessing steps and data splitting strategies through Git branches, ensuring reproducibility and project isolation. The original paper must be cited when using this dataset.
创建时间:
2026-04-23
原始信息汇总
GEOM-Drugs 数据集概述
基本信息
- 数据集名称: GEOM-Drugs (镜像版本)
- 许可证: CC-BY-4.0
- 标签: 化学、构象生成、分子
- 数据集地址: https://huggingface.co/datasets/raulsofia/geom_drugs
数据集描述
GEOM(分子几何集成)数据集包含数百万个分子构象,每个构象都带有能量和统计权重标注。本仓库托管的是“Drugs”子集,专注于具有实验数据的、尺寸接近真实药物的分子。
原始来源
- 原始论文: GEOM, energy-annotated molecular conformations for property prediction and molecular generation
- 原始GitHub: learningmatter-mit/geom
- 原始作者: Simon Axelrod 和 Rafael Gómez-Bombarelli
版本与分支说明
- 主分支 (main): 作为规范基础版本,包含基本未改动(或最小预处理)的原始数据集。
- 项目分支: 仓库使用Git分支存储项目特定的预处理和数据划分信息。
- 每个分支对应一个特定项目,预处理步骤在外部代码库中完整记录。
- 数据划分采用索引策略:分支存储轻量级索引数组,下游代码在运行时动态基于规范主文件进行子集划分,避免重复存储大量数据。
引用信息
使用本数据集时,必须引用原始作者:
bibtex @article{axelrod2022geom, title={GEOM, energy-annotated molecular conformations for property prediction and molecular generation}, author={Axelrod, Simon and G{o}mez-Bombarelli, Rafael}, journal={Scientific Data}, volume={9}, number={1}, pages={185}, year={2022}, publisher={Nature Publishing Group UK London} }
搜集汇总
数据集介绍
构建方式
GEOM-Drugs数据集源自GEOM(Geometric Ensemble Of Molecules)项目,旨在为机器学习研究提供大规模、能量标注的分子构象数据。其构建过程首先从多种公开数据库(如PubChem、ChEMBL等)中筛选出具有实验数据且分子量适中的类药小分子,随后利用先进的构象采样算法(CREST)生成大量稳定构象,并通过密度泛函理论(DFT)计算获取每个构象的相对能量与统计权重。该镜像版本在保留原始数据完整性的基础上,通过Git分支策略管理项目特定的预处理与数据划分,避免大规模重复存储,同时确保下游任务的可复现性。
特点
该数据集的核心特点在于规模宏大且信息丰富,包含数百万个类药分子的三维构象及对应的能量标签,为构象生成、分子性质预测与分子生成等领域提供了坚实的数据基础。其独特的版本管理机制允许不同项目在独立分支中灵活定义预处理步骤与数据划分索引,而无需修改原始主文件,从而实现了高效的存储与严格的实验隔离。此外,所有预处理变更均通过外部文档透明记录,保证了数据使用的可追溯性与研究结果的可靠性。
使用方法
研究者在利用该数据集时,应首先克隆仓库并检出所需的项目分支,每个分支中包含轻量级索引文件,用于从主数据文件中动态提取训练、验证和测试子集。推荐采用分子图神经网络或构象生成模型框架(如PyTorch Geometric、OpenMM等)直接读取构象坐标与能量信息。需注意,使用本数据集必须引用原始论文以尊重学术规范,且所有预处理配置的详细说明应参照对应分支的代码库文档进行操作。
背景与挑战
背景概述
分子构象生成是计算化学与药物设计领域的核心任务,其关键在于准确预测分子的三维结构以理解其物理化学性质与生物活性。GEOM-Drugs数据集由Simon Axelrod和Rafael Gómez-Bombarelli于2022年创建,隶属于麻省理工学院学习物质实验室,专注于包含实验数据的类药分子构象采样与密度泛函理论计算。该数据集通过提供数百万个能量注释的分子构象及其统计权重,显著推动了分子性质预测与分子生成模型的发展,为解决小分子构象空间探索这一长期难题奠定了数据基础,并成为相关领域广泛引用的基准资源。
当前挑战
数据集旨在应对类药分子构象空间复杂多样性的领域挑战,即如何从海量可能构象中识别关键结构并关联其能量分布。构建过程中面临的主要挑战包括:对数十万种不同分子系统性地生成低能构象并进行高精度量子化学计算,涉及巨大的计算资源与时间成本;确保采样方法覆盖构象空间的全面性与代表性,避免因局部极小值陷阱导致的偏差;以及处理数据索引与版本管理,以支持不同项目间灵活的子集划分与可重复性研究,如通过分支策略存储轻量级索引而非重复存储原始数据。
常用场景
经典使用场景
在计算化学与机器学习交叉领域中,GEOM-Drugs数据集被广泛用作分子构象生成与能量排序的基准测试平台。研究者利用其提供的数百万种能量标注且带有统计权重的药物分子三维构象,训练和评估深度生成模型(如变分自编码器、生成对抗网络或扩散模型)在从分子图直接预测稳定几何构象方面的表现。该数据集特别适合用于研究构象系综的覆盖率和能量排序的准确性,成为对比不同构象采样方法效力的黄金标准。
解决学术问题
GEOM-Drugs数据集的诞生系统性地解决了药物分子构象多样性量化与能量标注数据匮乏的核心瓶颈。在传统量子化学计算中,获取大量药物分子的精确构象能量分布需要高昂的计算成本,而该数据集通过事先完成的密度泛函理论计算,为机器学习模型提供了可靠的构象-能量对应关系。这使得研究人员可以聚焦于开发数据驱动的构象生成算法,并有效推动了对分子内非共价相互作用、旋转势垒以及柔性构象空间统计规律的深入理解。
衍生相关工作
GEOM-Drugs数据集催生了一系列具有影响力的衍生研究,包括但不限于ConfGF系列扩散模型、GeoMol等图神经网络构象生成架构,以及基于能量加权损失的构象排序优化方法。研究者还依托该数据开发了构象对比学习框架,用于学习不变性分子表征,并衍生出针对蛋白质-配体复合物构象预测的拓展工作。这些工作不仅验证了数据集的科学价值,还推动了分子动力学模拟增强采样方法与机器学习技术之间的深度融合。
以上内容由遇见数据集搜集并总结生成


