HuggingFaceBio/perturbation-bench
收藏Hugging Face2026-06-18 更新2026-06-14 收录
下载链接:
https://hf-mirror.com/datasets/HuggingFaceBio/perturbation-bench
下载链接
链接失效反馈官方服务:
资源简介:
一个用于评估DNA基础模型的序列级别扰动任务基准。每个任务呈现基因组序列对——一个是真实的(未扰动),另一个是结构上被改变的——并询问模型是否对原始序列分配更高的对数似然。**指标**:成对判别准确率 = `mean(LL(原始) > LL(扰动后))`。
A benchmark of **sequence-level perturbation tasks** for evaluating DNA foundation models. Each task presents pairs of genomic sequences — one real (unperturbed) and one structurally altered — and asks whether the model assigns higher log-likelihood to the original. **Metric**: pairwise discrimination accuracy = `mean(LL(original) > LL(perturbed))`
提供机构:
HuggingFaceBio搜集汇总
数据集介绍
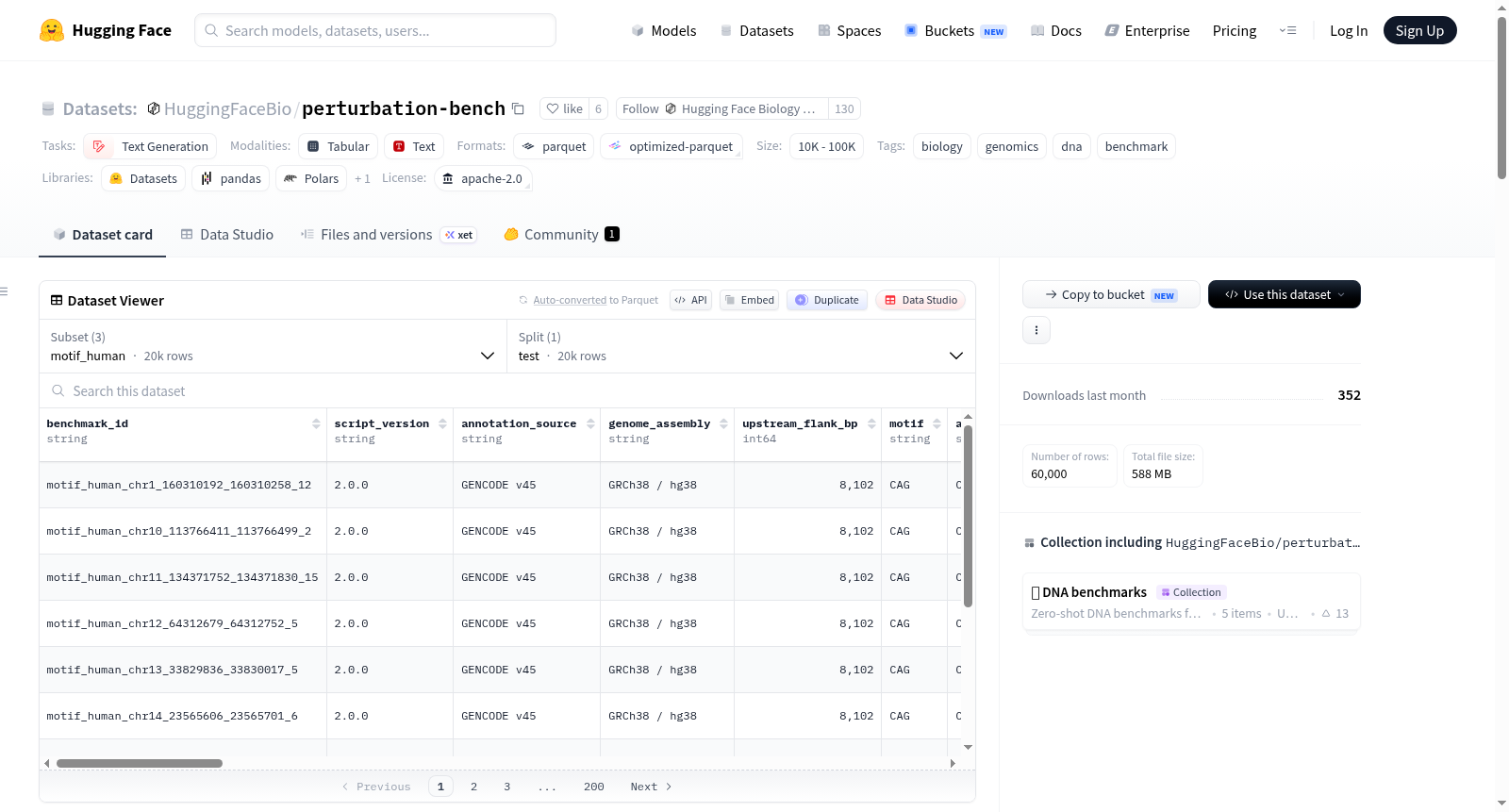
构建方式
Perturbation Bench 数据集旨在为评估DNA基础模型在序列层面扰动任务上的性能而构建。其构建核心在于生成成对的基因组序列:一条为真实未扰动的原始序列,另一条则经过人工设计的结构改变。具体而言,数据集包含三个独立的配置:syn_human和syn_mouse,分别针对人类和小鼠,通过将真实CDS区域内的密码子替换为目标物种中最高频的同义密码子来模拟同义替换扰动,同时保持氨基酸序列不变且上下游侧翼序列完整;motif_human则模拟病理性的三核苷酸重复扩增,通过在CDS外显子内的一段30 bp区域插入连续的CAG三联体序列。所有扰动均保持序列长度与阅读框不变,最终窗口大小为8192 bp,以确保模型评估的准确性。
特点
该数据集具备三项突出特点。其一,任务设计具有明确的生物学动机:同义密码子替换任务评估模型对自然密码子使用偏好的判断能力,而CAG重复插入任务则直接模拟了亨廷顿病等聚谷氨酰胺疾病的遗传变异背景,使评估结果更贴近真实生物学问题。其二,覆盖物种广泛,包含人类与小鼠的多达数万个独特基因,且每个配置均提供20000个测试样本,统计能力充足。其三,数据注释丰富,每条样本均附带基因组坐标、转录本与基因标识、CDS边界、扰动补丁位置及密码子改变比例等元信息,便于深入分析与可视化。
使用方法
数据集的使用流程简洁高效,依托HuggingFace的datasets库即可一键加载。用户可通过load_dataset函数分别指定三个配置名称——syn_human、syn_mouse或motif_human——并选择test分片来获取完整的测试集。评估指标为逐对判别准确率,即比较模型对原始序列与扰动序列的对数似然值,正确判别原始序列为更优的比率。此外,项目还提供了现成的评测脚本,支持Carbon、GENERator及Evo2等主流DNA模型,通过命令行参数指定任务与模型即可复现基准结果,极大降低了研究者开展对比实验的门槛。
背景与挑战
背景概述
在基因组学与深度学习交叉领域,DNA基础模型的评估一直是关键瓶颈,亟需能够精准衡量模型对序列生物学特性理解能力的基准测试。Perturbation Bench数据集由HuggingFace团队联合多位研究人员于2026年构建,旨在通过序列层面的扰动任务来系统评估DNA基础模型的性能。该数据集聚焦于两大核心生物学问题:同义密码子使用的偏好性以及病理性的三核苷酸重复扩增。通过设计syn_human、syn_mouse和motif_human三个子任务,覆盖人类和小鼠基因组,每个任务包含20000个序列对,要求模型判定真实序列与人工扰动序列之间的似然度高低。这一基准填补了现有DNA模型评估中缺乏结构化扰动测试的空白,为Evo2、Carbon等前沿基因组语言模型提供了标准化的性能度量工具,推动了计算基因组学评估范式的进步。
当前挑战
Perturbation Bench所解决的领域核心挑战在于,DNA基础模型不仅要捕捉序列的统计规律,还需理解其背后的生物学含义,如同义密码子使用偏向和重复序列的病理效应。构建过程中的挑战体现在三个层面:首先,同义替换任务需要精准保持氨基酸序列不变的同时改变密码子使用频率,这要求对GENCODE注释和CoCoPUTs密码子频率数据的高质量整合;其次,人类和小鼠基因组的同义替换需分别处理GRCh38和GRCm39两个不同版本的组装,确保跨物种的基准一致性;第三,CAG重复插入任务模拟亨廷顿病等三核苷酸重复疾病,必须在保持读码框和序列长度的前提下精确替换30个碱基的编码区片段,这涉及对基因外显子相位和CDS边界的复杂处理,任何一个坐标偏差都会导致生物学假象,从而影响评估的可靠性。
常用场景
经典使用场景
在基因组学与计算生物学交叉领域,Perturbation Bench基准数据集专为评估DNA基础模型对序列扰动的敏感性而设计。其核心任务聚焦于成对序列判别——给定一条真实基因组序列与一条结构改变后的序列,模型需准确判断原始序列是否具有更高的对数似然值。该数据集精心构建了三类扰动场景:同义密码子替换(涵盖人类与小鼠)、CAG重复序列插入(模拟多聚谷氨酰胺疾病的病理特征)。每一场景均包含20,000个测试样本,窗口长度固定为8,192个碱基对,确保了序列上下文的完整性与评估的统计稳健性。
解决学术问题
该数据集系统性地解决了DNA基础模型在序列级扰动判别能力上的评估缺失问题。传统基准往往侧重于序列的简单分类或生成任务,未能有效衡量模型对微妙遗传变异的感知精度。Perturbation Bench通过构建生理性与病理性扰动事件,直接检验模型是否具备捕捉天然密码子偏好性、识别致病性重复序列扩张等关键生物学特征的内在能力。其意义在于为模型开发者提供了一种标准化、可复现的评估范式,推动DNA语言模型从粗粒度表征向真正理解基因组编码规则的精细化方向演进,进而提升模型在功能基因组学预测任务中的科学可靠性。
衍生相关工作
围绕Perturbation Bench已衍生出一系列标志性研究成果。Carbon模型团队在其发布中首次将本数据集作为核心评估基准,系统展示了模型在同义密码子偏好性判别与三核苷酸重复扩张检测上的卓越性能。通过对该数据的深度分析,研究者进一步探讨了不同DNA语言模型在序列上下文编码、突变语义理解等方面的本质差异,催生了关于如何融合进化约束、密码子频率等先验知识来提升模型鲁棒性的多项后续工作。该数据集的引入实质性地推动了DNA基础模型评估体系从孤立任务向多维度扰动情境的范式转变。
以上内容由遇见数据集搜集并总结生成


